

Current and future therapies for inherited cholestatic liver diseases
- PMID: 28223721
- PMCID: PMC5296193
- DOI: 10.3748/wjg.v23.i5.763
Current and future therapies for inherited cholestatic liver diseases
Abstract
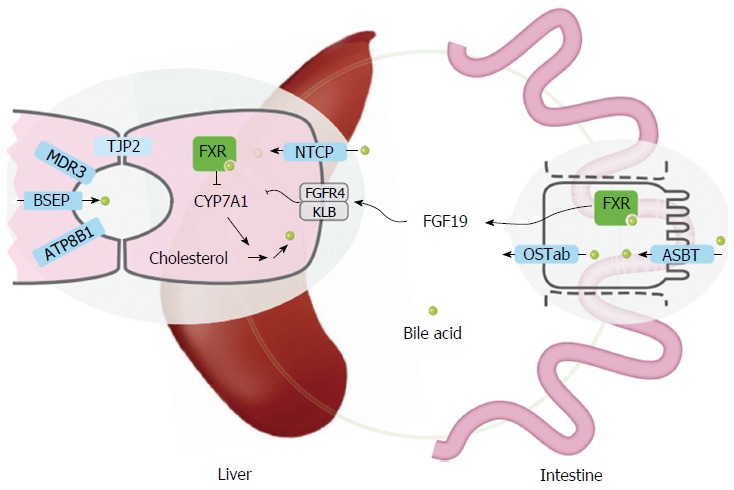
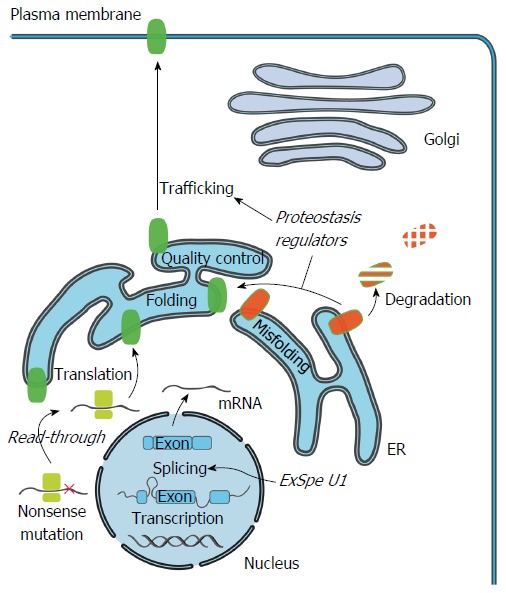
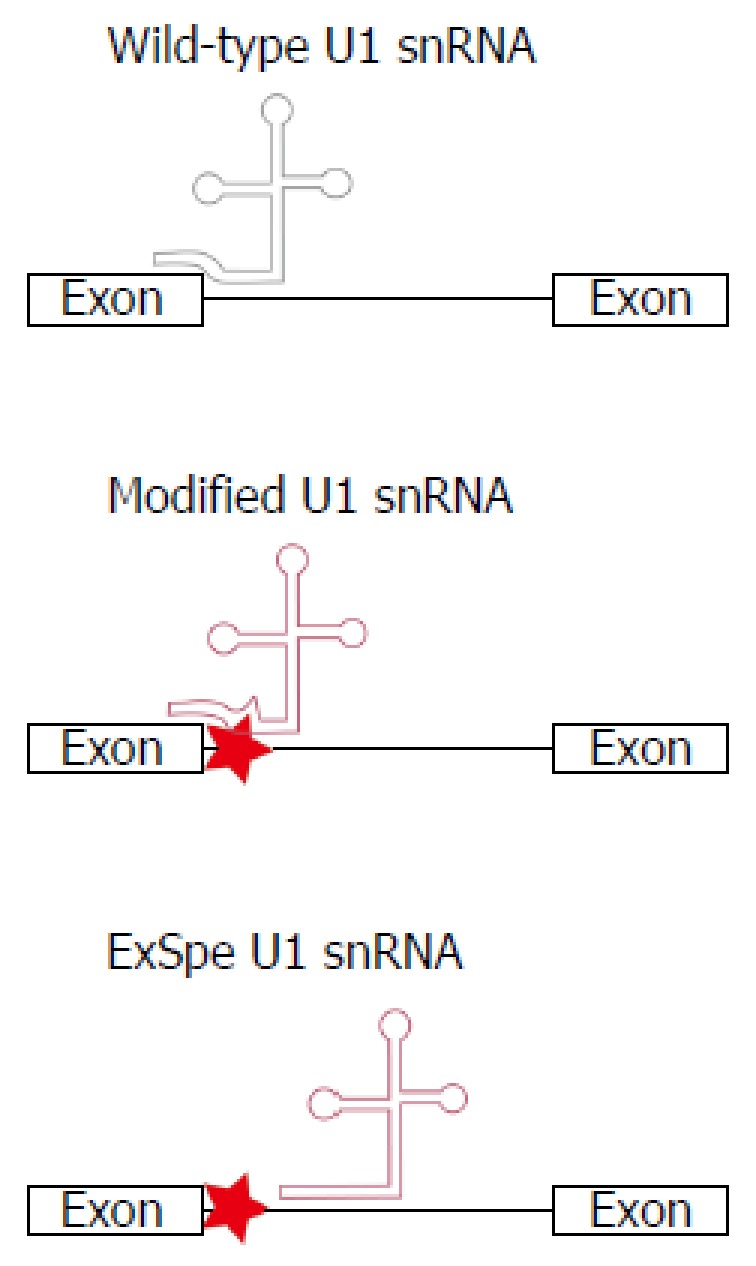
Familial intrahepatic cholestasis (FIC) comprises a group of rare cholestatic liver diseases associated with canalicular transport defects resulting predominantly from mutations in ATP8B1, ABCB11 and ABCB4. Phenotypes range from benign recurrent intrahepatic cholestasis (BRIC), associated with recurrent cholestatic attacks, to progressive FIC (PFIC). Patients often suffer from severe pruritus and eventually progressive cholestasis results in liver failure. Currently, first-line treatment includes ursodeoxycholic acid in patients with ABCB4 deficiency (PFIC3) and partial biliary diversion in patients with ATP8B1 or ABCB11 deficiency (PFIC1 and PFIC2). When treatment fails, liver transplantation is needed which is associated with complications like rejection, post-transplant hepatic steatosis and recurrence of disease. Therefore, the need for more and better therapies for this group of chronic diseases remains. Here, we discuss new symptomatic treatment options like total biliary diversion, pharmacological diversion of bile acids and hepatocyte transplantation. Furthermore, we focus on emerging mutation-targeted therapeutic strategies, providing an outlook for future personalized treatment for inherited cholestatic liver diseases.
Keywords: ABCB11; ABCB4; ATP8B1; Biliary diversion; Familial intrahepatic cholestasis; Inherited liver disease; Mutation-targeted therapy; Personalized treatment; Progressive familial intrahepatic cholestasis.
Conflict of interest statement
Conflict-of-interest statement: Authors declare no conflict of interest for this article.
Figures
Similar articles
-
Progressive familial intrahepatic cholestasis.Clin Res Hepatol Gastroenterol. 2012 Sep;36 Suppl 1:S26-35. doi: 10.1016/S2210-7401(12)70018-9. Clin Res Hepatol Gastroenterol. 2012. PMID: 23141890 Review.
-
Familial cholestasis: progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancy.Best Pract Res Clin Gastroenterol. 2010 Oct;24(5):541-53. doi: 10.1016/j.bpg.2010.07.010. Best Pract Res Clin Gastroenterol. 2010. PMID: 20955958 Review.
-
ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history.Hepatology. 2010 May;51(5):1645-55. doi: 10.1002/hep.23539. Hepatology. 2010. PMID: 20232290
-
ATP8B1, ABCB11, and ABCB4 Genes Defects: Novel Mutations Associated with Cholestasis with Different Phenotypes and Outcomes.J Pediatr. 2021 Sep;236:113-123.e2. doi: 10.1016/j.jpeds.2021.04.040. Epub 2021 Apr 27. J Pediatr. 2021. PMID: 33915153
-
Heterozygous mutations of ATP8B1, ABCB11 and ABCB4 cause mild forms of Progressive Familial Intrahepatic Cholestasis in a pediatric cohort.Gastroenterol Hepatol. 2022 Oct;45(8):585-592. doi: 10.1016/j.gastrohep.2021.12.005. Epub 2021 Dec 20. Gastroenterol Hepatol. 2022. PMID: 34942279 English, Spanish.
Cited by
-
ABCB4 Mutations in Adults Cause a Spectrum Cholestatic Disorder Histologically Distinct from Other Biliary Disease.Dig Dis Sci. 2022 Dec;67(12):5551-5561. doi: 10.1007/s10620-022-07416-9. Epub 2022 Mar 14. Dig Dis Sci. 2022. PMID: 35288833
-
Biliary atresia combined with progressive familial intrahepatic cholestasis type 3: A case report and review of the literature.Medicine (Baltimore). 2019 May;98(19):e15593. doi: 10.1097/MD.0000000000015593. Medicine (Baltimore). 2019. PMID: 31083246 Free PMC article. Review.
-
Effect of nalfurafine hydrochloride in patients with chronic liver disease with refractory pruritus on sleep disorders: a study protocol for single-arm, prospective, interventional study.BMJ Open Gastroenterol. 2017 Sep 29;4(1):e000177. doi: 10.1136/bmjgast-2017-000177. eCollection 2017. BMJ Open Gastroenterol. 2017. PMID: 29104757 Free PMC article.
-
Bile acid indices as biomarkers for liver diseases I: Diagnostic markers.World J Hepatol. 2021 Apr 27;13(4):433-455. doi: 10.4254/wjh.v13.i4.433. World J Hepatol. 2021. PMID: 33959226 Free PMC article.
-
New Insights in Genetic Cholestasis: From Molecular Mechanisms to Clinical Implications.Can J Gastroenterol Hepatol. 2018 Jul 26;2018:2313675. doi: 10.1155/2018/2313675. eCollection 2018. Can J Gastroenterol Hepatol. 2018. PMID: 30148122 Free PMC article. Review.
References
-
- Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. - PubMed
-
- Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. - PubMed
-
- Gordo-Gilart R, Andueza S, Hierro L, Martínez-Fernández P, D’Agostino D, Jara P, Alvarez L. Functional analysis of ABCB4 mutations relates clinical outcomes of progressive familial intrahepatic cholestasis type 3 to the degree of MDR3 floppase activity. Gut. 2015;64:147–155. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
