

Fibrodysplasia ossificans progressiva: clinical and genetic aspects
- PMID: 22133093
- PMCID: PMC3253727
- DOI: 10.1186/1750-1172-6-80
Fibrodysplasia ossificans progressiva: clinical and genetic aspects
Abstract
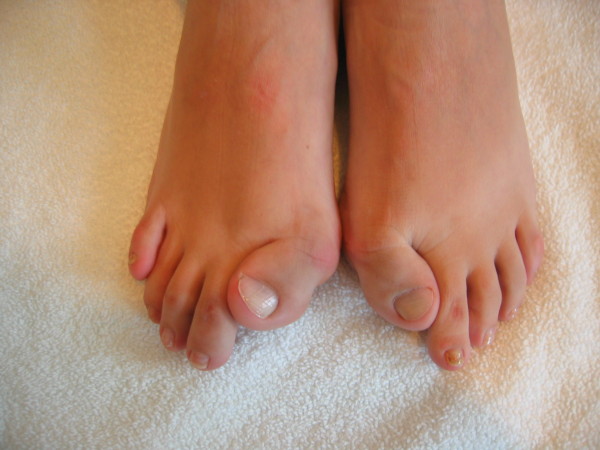
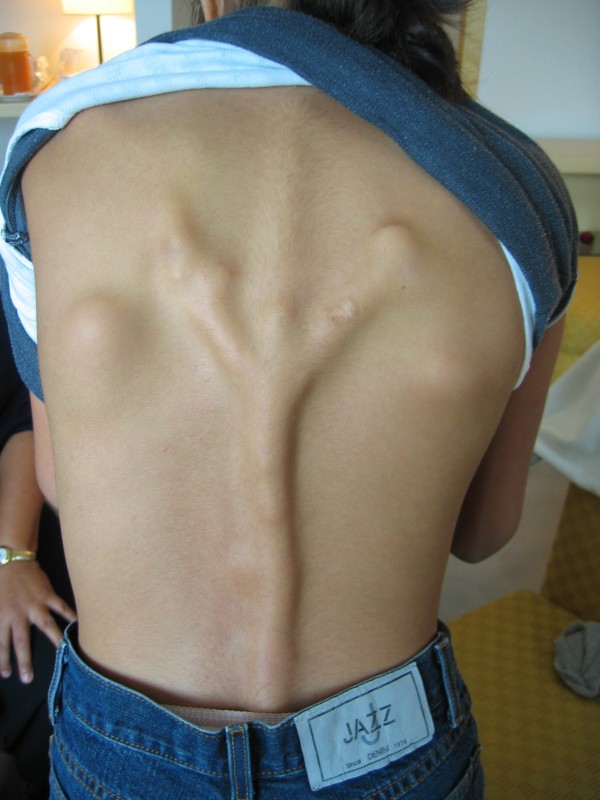
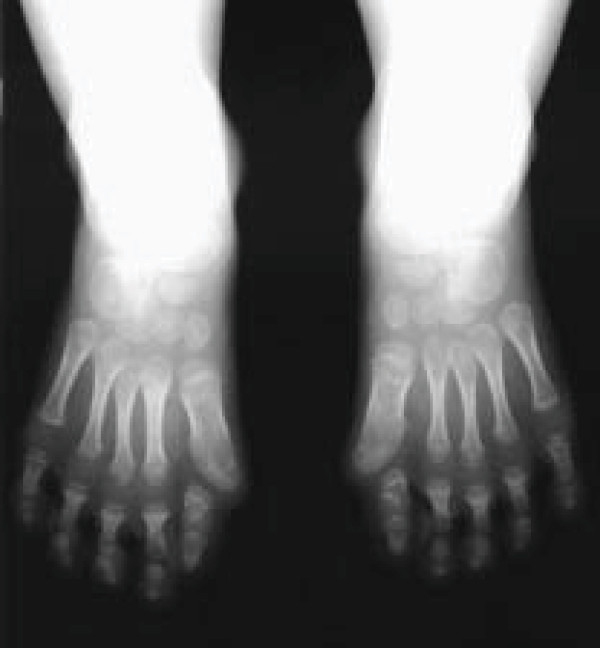
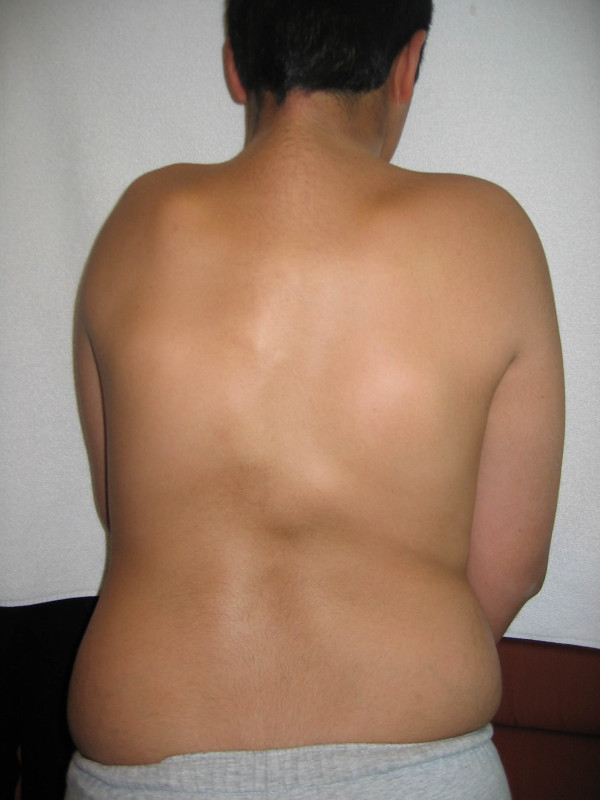
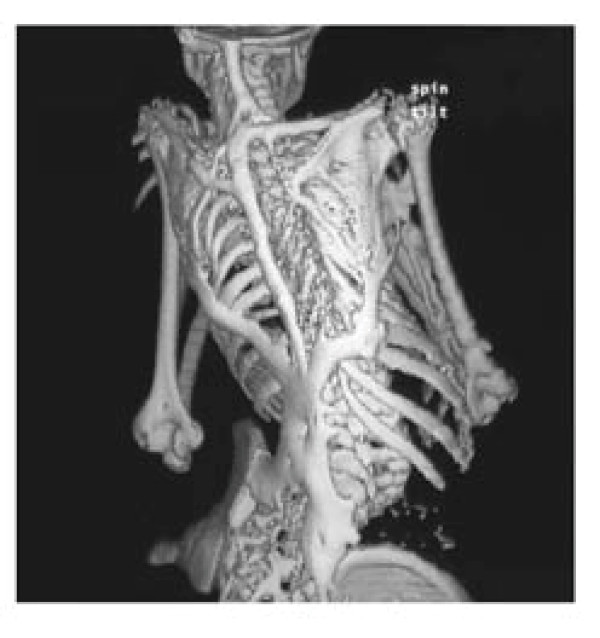
Fibrodysplasia ossificans progressiva (FOP) is a severely disabling heritable disorder of connective tissue characterized by congenital malformations of the great toes and progressive heterotopic ossification that forms qualitatively normal bone in characteristic extraskeletal sites. The worldwide prevalence is approximately 1/2,000,000. There is no ethnic, racial, gender, or geographic predilection to FOP. Children who have FOP appear normal at birth except for congenital malformations of the great toes. During the first decade of life, sporadic episodes of painful soft tissue swellings (flare-ups) occur which are often precipitated by soft tissue injury, intramuscular injections, viral infection, muscular stretching, falls or fatigue. These flare-ups transform skeletal muscles, tendons, ligaments, fascia, and aponeuroses into heterotopic bone, rendering movement impossible. Patients with atypical forms of FOP have been described. They either present with the classic features of FOP plus one or more atypical features [FOP plus], or present with major variations in one or both of the two classic defining features of FOP [FOP variants]. Classic FOP is caused by a recurrent activating mutation (617G>A; R206H) in the gene ACVR1/ALK2 encoding Activin A receptor type I/Activin-like kinase 2, a bone morphogenetic protein (BMP) type I receptor. Atypical FOP patients also have heterozygous ACVR1 missense mutations in conserved amino acids. The diagnosis of FOP is made by clinical evaluation. Confirmatory genetic testing is available. Differential diagnosis includes progressive osseous heteroplasia, osteosarcoma, lymphedema, soft tissue sarcoma, desmoid tumors, aggressive juvenile fibromatosis, and non-hereditary (acquired) heterotopic ossification. Although most cases of FOP are sporadic (noninherited mutations), a small number of inherited FOP cases show germline transmission in an autosomal dominant pattern. At present, there is no definitive treatment, but a brief 4-day course of high-dose corticosteroids, started within the first 24 hours of a flare-up, may help reduce the intense inflammation and tissue edema seen in the early stages of the disease. Preventative management is based on prophylactic measures against falls, respiratory decline, and viral infections. The median lifespan is approximately 40 years of age. Most patients are wheelchair-bound by the end of the second decade of life and commonly die of complications of thoracic insufficiency syndrome.
Figures
Similar articles
-
Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1.Hum Mutat. 2009 Mar;30(3):379-90. doi: 10.1002/humu.20868. Hum Mutat. 2009. PMID: 19085907 Free PMC article.
-
Fibrodysplasia ossificans progressiva (FOP): watch the great toes!Eur J Pediatr. 2010 Nov;169(11):1417-21. doi: 10.1007/s00431-010-1232-5. Epub 2010 Jun 26. Eur J Pediatr. 2010. PMID: 20577760 Free PMC article.
-
Fibrodysplasia ossificans progressiva.Best Pract Res Clin Rheumatol. 2008 Mar;22(1):191-205. doi: 10.1016/j.berh.2007.11.007. Best Pract Res Clin Rheumatol. 2008. PMID: 18328989 Free PMC article. Review.
-
Fibrodysplasia Ossificans Progressiva: A Challenging Diagnosis.Genes (Basel). 2021 Jul 30;12(8):1187. doi: 10.3390/genes12081187. Genes (Basel). 2021. PMID: 34440363 Free PMC article.
-
The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva.Bone. 2018 Apr;109:210-217. doi: 10.1016/j.bone.2017.06.011. Epub 2017 Jun 16. Bone. 2018. PMID: 28629737 Free PMC article. Review.
Cited by
-
Fibrodysplasia ossificans progressiva (FOP) presenting as a rapidly growing non-calcified neck mass.J Radiol Case Rep. 2021 May 31;15(5):10-16. doi: 10.3941/jrcr.v15i5.4103. eCollection 2021 May. J Radiol Case Rep. 2021. PMID: 34276874 Free PMC article.
-
Transcriptomic Differences Underlying the Activin-A Induced Large Osteoclast Formation in Both Healthy Control and Fibrodysplasia Ossificans Progressiva Osteoclasts.Int J Mol Sci. 2023 Apr 6;24(7):6822. doi: 10.3390/ijms24076822. Int J Mol Sci. 2023. PMID: 37047804 Free PMC article.
-
Application of human induced pluripotent stem cells to model fibrodysplasia ossificans progressiva.Bone. 2018 Apr;109:162-167. doi: 10.1016/j.bone.2017.07.003. Epub 2017 Jul 14. Bone. 2018. PMID: 28716551 Free PMC article. Review.
-
The stone women-Myositis ossificans Progressiva.J Orthop Case Rep. 2015 Oct-Dec;5(4):10-3. doi: 10.13107/jocr.2250-0685.333. J Orthop Case Rep. 2015. PMID: 27299087 Free PMC article.
-
Signaling pathways regulating cartilage growth plate formation and activity.Semin Cell Dev Biol. 2017 Feb;62:3-15. doi: 10.1016/j.semcdb.2016.07.008. Epub 2016 Jul 11. Semin Cell Dev Biol. 2017. PMID: 27418125 Free PMC article. Review.
References
-
- OMIM. Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/omim
-
- Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:201–204. doi: 10.1385/BMM:3:3-4:201. - DOI
-
- Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age-and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1994. pp. 243–248. - PubMed
-
- Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am. 1993;75:215–219. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
