

High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension
- PMID: 16728714
- PMCID: PMC2648061
- DOI: 10.1164/rccm.200602-165OC
High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension
Abstract
Rationale: Previous studies have shown that approximately 55% of patients with familial pulmonary arterial hypertension (FPAH) have BMPR2 coding sequence mutations. However, direct sequencing does not detect other types of heterozygous mutations, such as exonic deletions/duplications.
Objective: To estimate the frequency of BMPR2 exonic deletions/duplications in FPAH.
Methods: BMPR2 mRNA from lymphoblastoid cell lines of 30 families with PAH and 14 patients with idiopathic PAH (IPAH) was subjected to reverse transcriptase-polymerase chain reaction (RT-PCR) and sequencing. Sequencing of genomic DNA was used to identify point mutations in splice donor/acceptor sites. Multiplex ligation-dependent probe amplification (MLPA) was used to detect exonic deletions/duplications with verification by real-time PCR.
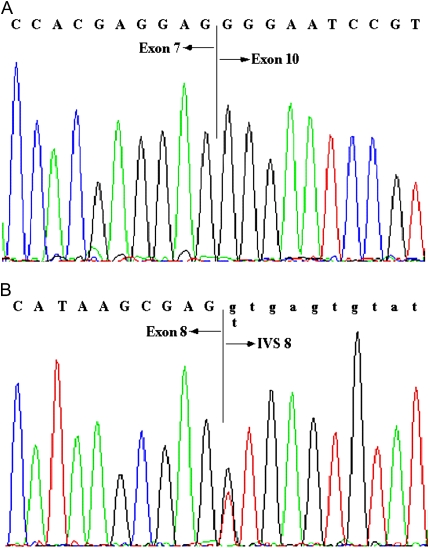
Measurements and main results: Eleven (37%) patients with FPAH had abnormally sized RT-PCR products. Four of the 11 patients were found to have splice-site mutations resulting in aberrant splicing, and exonic deletions/duplications were detected in the remaining seven patients. MLPA identified three deletions/duplications that were not detectable by RT-PCR. Coding sequence point mutations were identified in 11 of 30 (37%) patients. Mutations were identified in 21 of 30 (70%) patients with FPAH, with 10 of 21 mutations (48%) being exonic deletions/duplications. Two of 14 (14%) patients with IPAH exhibited BMPR2 point mutations, whereas none showed exonic deletions/duplications.
Conclusions: Our study indicates that BMPR2 exonic deletions/duplications in patients with FPAH account for a significant proportion of mutations (48%) that until now have not been screened for. Because the complementary approach used in this study is rapid and cost effective, screening for BMPR2 deletions/duplications by MLPA and real-time PCR should accompany direct sequencing in all PAH testing.
Figures





Similar articles
-
A novel mutation in the BMPR2 gene in familial pulmonary arterial hypertension.Chin Med J (Engl). 2008 Mar 5;121(5):399-404. Chin Med J (Engl). 2008. PMID: 18364108
-
BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension.Hum Mutat. 2006 Feb;27(2):212-3. doi: 10.1002/humu.9398. Hum Mutat. 2006. PMID: 16429403
-
Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension.Hum Mutat. 2006 Feb;27(2):121-32. doi: 10.1002/humu.20285. Hum Mutat. 2006. PMID: 16429395
-
Genes and pulmonary arterial hypertension.Respiration. 2007;74(2):123-32. doi: 10.1159/000098818. Respiration. 2007. PMID: 17318011 Review.
-
Genetic basis of pulmonary arterial hypertension: current understanding and future directions.J Am Coll Cardiol. 2004 Jun 16;43(12 Suppl S):33S-39S. doi: 10.1016/j.jacc.2004.02.028. J Am Coll Cardiol. 2004. PMID: 15194176 Review.
Cited by
-
Chronic allergic inflammation causes vascular remodeling and pulmonary hypertension in BMPR2 hypomorph and wild-type mice.PLoS One. 2012;7(3):e32468. doi: 10.1371/journal.pone.0032468. Epub 2012 Mar 9. PLoS One. 2012. PMID: 22427841 Free PMC article.
-
Identification of a low frequency missense mutation in MUC6 contributing to pulmonary artery hypertension by whole-exome sequencing.Pulm Circ. 2018 Jul-Sep;8(3):2045894018794374. doi: 10.1177/2045894018794374. Epub 2018 Jul 26. Pulm Circ. 2018. PMID: 30047301 Free PMC article.
-
Transcripts from a novel BMPR2 termination mutation escape nonsense mediated decay by downstream translation re-initiation: implications for treating pulmonary hypertension.Clin Genet. 2010 Mar;77(3):280-6. doi: 10.1111/j.1399-0004.2009.01311.x. Epub 2010 Jan 20. Clin Genet. 2010. PMID: 20095988 Free PMC article.
-
Isolated growth hormone deficiency type II caused by a point mutation that alters both splice site strength and splicing enhancer function.Clin Genet. 2008 Dec;74(6):539-45. doi: 10.1111/j.1399-0004.2008.01042.x. Epub 2008 Jun 11. Clin Genet. 2008. PMID: 18554279 Free PMC article.
-
BMPR2 mutations and endothelial dysfunction in pulmonary arterial hypertension (2017 Grover Conference Series).Pulm Circ. 2018 Apr-Jun;8(2):2045894018765840. doi: 10.1177/2045894018765840. Epub 2018 Mar 9. Pulm Circ. 2018. PMID: 29521190 Free PMC article.
References
-
- Torbicki A, Kurzyna M. Pulmonary arterial hypertension: evaluation of the newly diagnosed patient. Semin Respir Crit Care Med 2005;26:372–378. - PubMed
-
- Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004;43:5S–12S. - PubMed
-
- Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 2004;43:25S–32S. - PubMed
-
- Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med 1987;107:216–223. - PubMed
-
- Chakinala MM. Changing the prognosis of pulmonary arterial hypertension: impact of medical therapy. Semin Respir Crit Care Med 2005;26: 409–416. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
