

Pseudohypoparathyroidism: complex disease variants with unfortunate names
- PMID: 37965945
- PMCID: PMC10843601
- DOI: 10.1530/JME-23-0104
Pseudohypoparathyroidism: complex disease variants with unfortunate names
Abstract
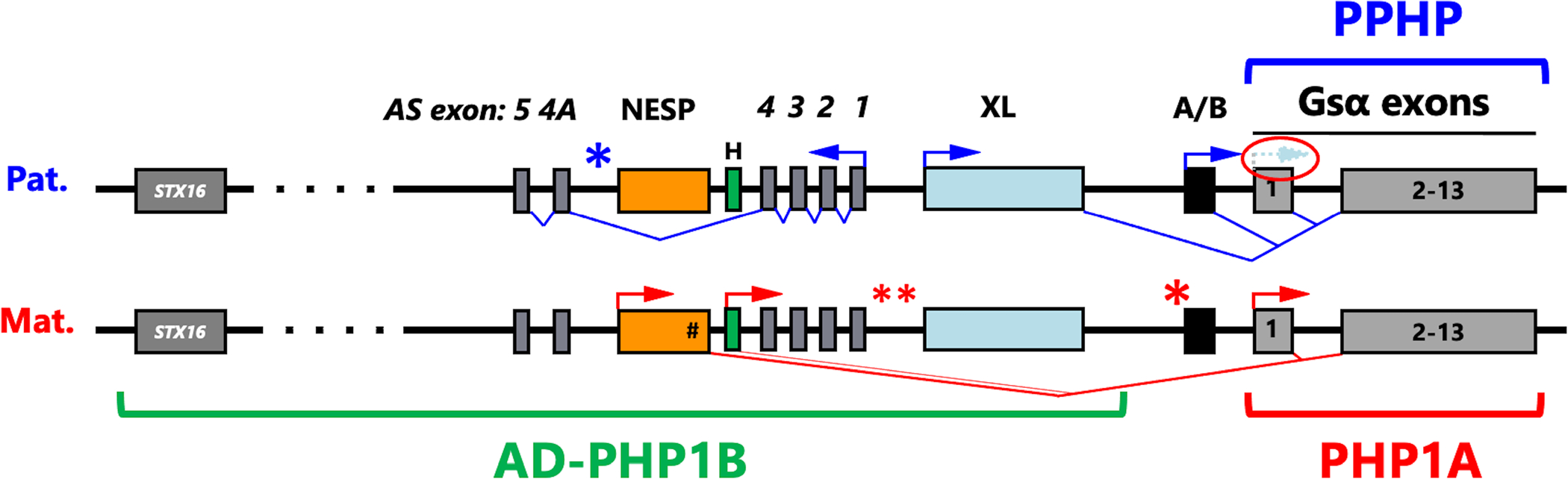
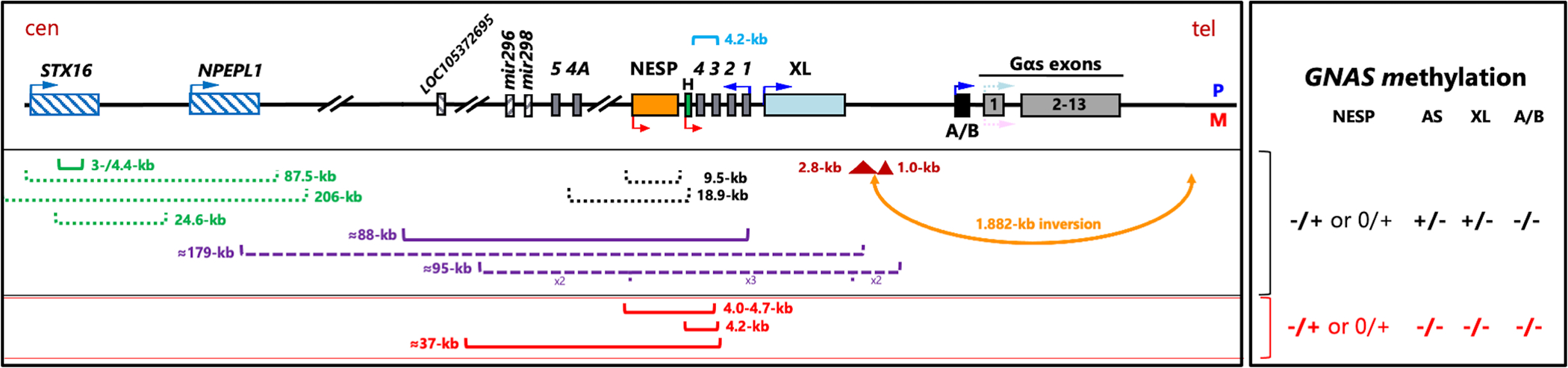
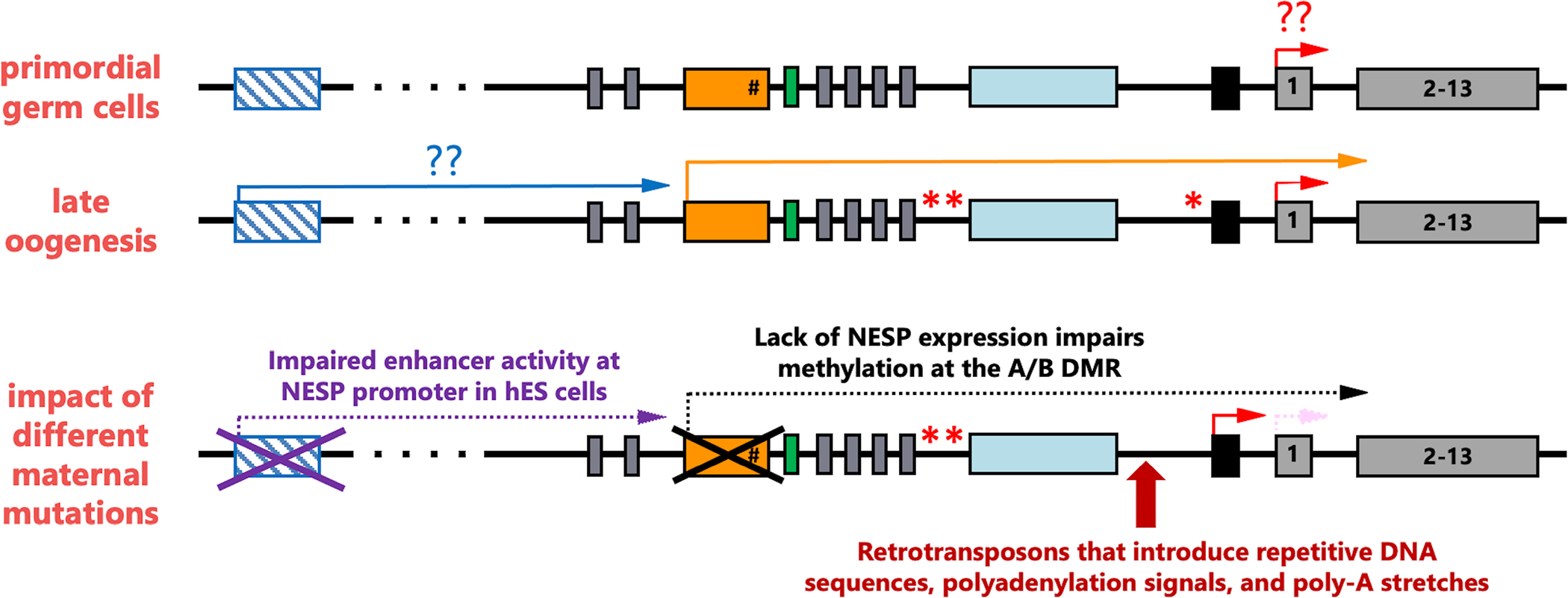
Several human disorders are caused by genetic or epigenetic changes involving the GNAS locus on chromosome 20q13.3 that encodes the alpha-subunit of the stimulatory G protein (Gsα) and several splice variants thereof. Thus, pseudohypoparathyroidism type Ia (PHP1A) is caused by heterozygous inactivating mutations involving the maternal GNAS exons 1-13 resulting in characteristic abnormalities referred to as Albright's hereditary osteodystrophy (AHO) that are associated with resistance to several agonist ligands, particularly to parathyroid hormone (PTH), thereby leading to hypocalcemia and hyperphosphatemia. GNAS mutations involving the paternal Gsα exons also cause most of these AHO features, but without evidence for hormonal resistance, hence the term pseudopseudohypoparathyroidism (PPHP). Autosomal dominant pseudohypoparathyroidism type Ib (PHP1B) due to maternal GNAS or STX16 mutations (deletions, duplications, insertions, and inversions) is associated with epigenetic changes at one or several differentially methylated regions (DMRs) within GNAS. Unlike the inactivating Gsα mutations that cause PHP1A and PPHP, hormonal resistance is caused in all PHP1B variants by impaired Gsα expression due to loss of methylation at GNAS exon A/B, which can be associated in some familial cases with epigenetic changes at the other maternal GNAS DMRs. The genetic defect(s) responsible for sporadic PHP1B, the most frequent variant of this disorder, remain(s) unknown for the majority of patients. However, characteristic epigenetic GNAS changes can be readily detected that include a gain of methylation at the neuroendocrine secretory protein (NESP) DMR. Multiple genetic or epigenetic GNAS abnormalities can thus impair Gsα function or expression, consequently leading to inadequate cAMP-dependent signaling events downstream of various Gsα-coupled receptors.
Keywords: GNAS; Gs-alpha; PTH; STX16; TSH; cAMP; calcium; epigenetics; parent-specific GNAS methylation; phosphate; pseudohypoparathyroidism.
Conflict of interest statement
There is no conflict of interest.
Figures
Similar articles
-
Molecular Definition of Pseudohypoparathyroidism Variants.J Clin Endocrinol Metab. 2021 May 13;106(6):1541-1552. doi: 10.1210/clinem/dgab060. J Clin Endocrinol Metab. 2021. PMID: 33529330 Free PMC article. Review.
-
Pseudohypoparathyroidism: one gene, several syndromes.J Endocrinol Invest. 2017 Apr;40(4):347-356. doi: 10.1007/s40618-016-0588-4. Epub 2016 Dec 19. J Endocrinol Invest. 2017. PMID: 27995443 Review.
-
Analysis of Multiple Families With Single Individuals Affected by Pseudohypoparathyroidism Type Ib (PHP1B) Reveals Only One Novel Maternally Inherited GNAS Deletion.J Bone Miner Res. 2016 Apr;31(4):796-805. doi: 10.1002/jbmr.2731. Epub 2015 Nov 14. J Bone Miner Res. 2016. PMID: 26479409 Free PMC article. Clinical Trial.
-
A Large Inversion Involving GNAS Exon A/B and All Exons Encoding Gsα Is Associated With Autosomal Dominant Pseudohypoparathyroidism Type Ib (PHP1B).J Bone Miner Res. 2017 Apr;32(4):776-783. doi: 10.1002/jbmr.3083. Epub 2017 Feb 24. J Bone Miner Res. 2017. PMID: 28084650 Free PMC article.
-
The GNAS locus and pseudohypoparathyroidism.Adv Exp Med Biol. 2008;626:27-40. doi: 10.1007/978-0-387-77576-0_3. Adv Exp Med Biol. 2008. PMID: 18372789 Review.
Cited by
-
A novel GNAS mutation in pseudohypoparathyroidism type 1a with articular flexion deformity: A case report.Open Life Sci. 2024 Jul 24;19(1):20220918. doi: 10.1515/biol-2022-0918. eCollection 2024. Open Life Sci. 2024. PMID: 39071491 Free PMC article.
-
Primary ovarian insufficiency: update on clinical and genetic findings.Front Endocrinol (Lausanne). 2024 Sep 26;15:1464803. doi: 10.3389/fendo.2024.1464803. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 39391877 Free PMC article. Review.
References
-
- ALBRIGHT F, BURNETT CH, SMITH PH & PARSON W 1942. Pseudohypoparathyroidism - an example of “Seabright-Bantam syndrome”. Endocrinology, 30, 922–932.
-
- ALBRIGHT F, FORBES AP & HENNEMAN PH 1952. Pseudo-pseudohypoparathyroidism. Trans. Assoc. Am. Physicians, 65, 337–350. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
