

Free sialic acid storage disorder: Progress and promise
- PMID: 33862140
- PMCID: PMC8175077
- DOI: 10.1016/j.neulet.2021.135896
Free sialic acid storage disorder: Progress and promise
Abstract
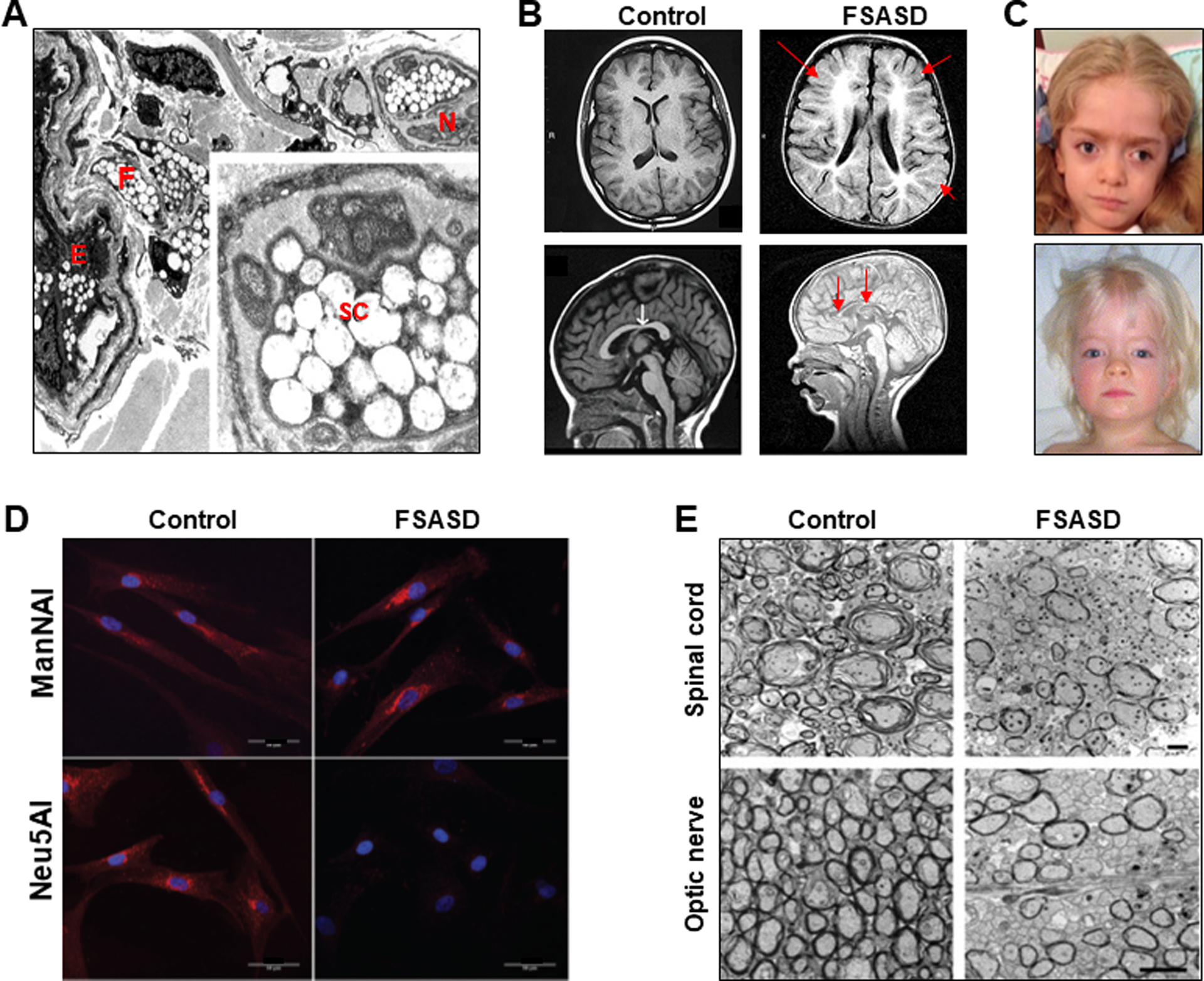
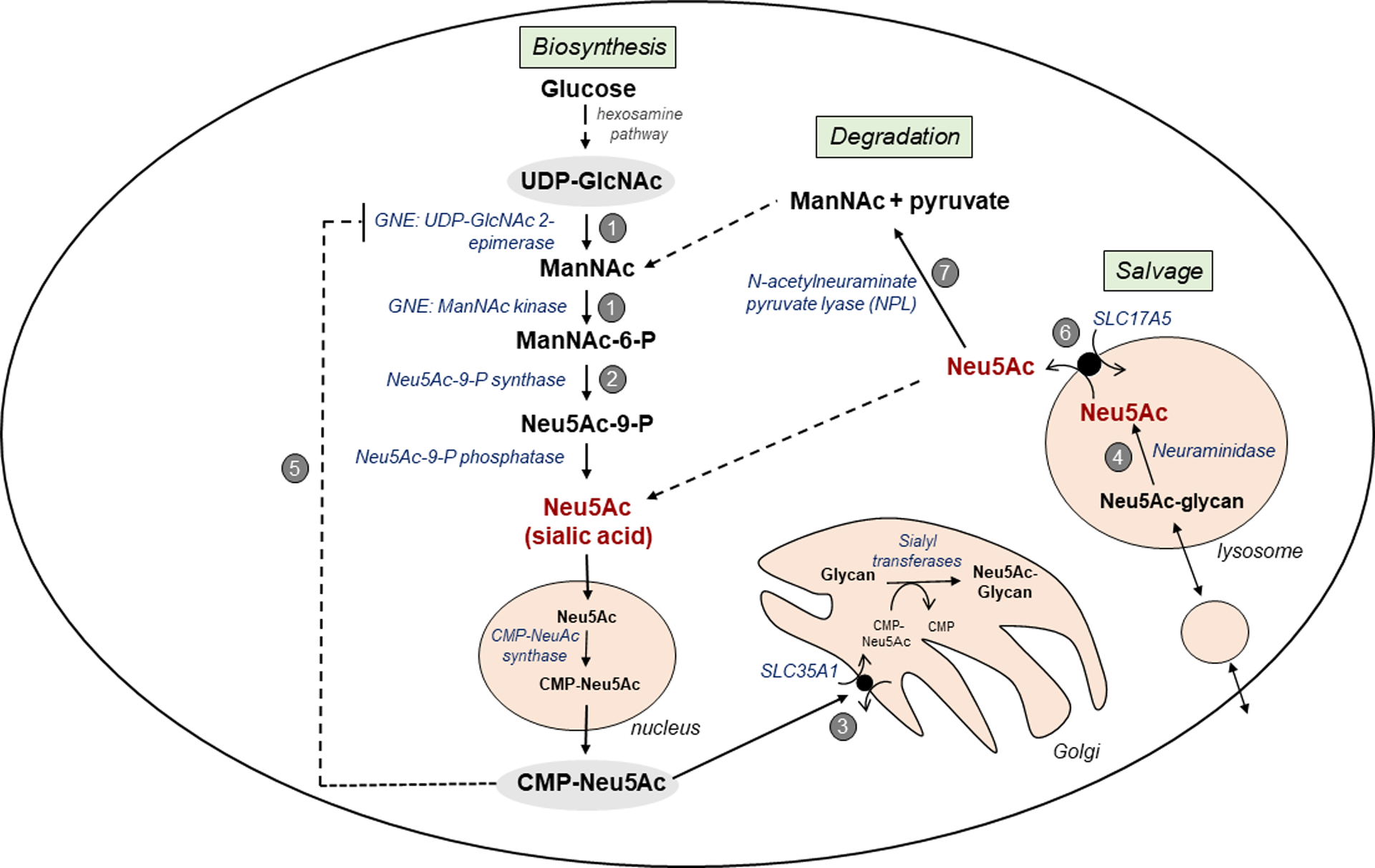
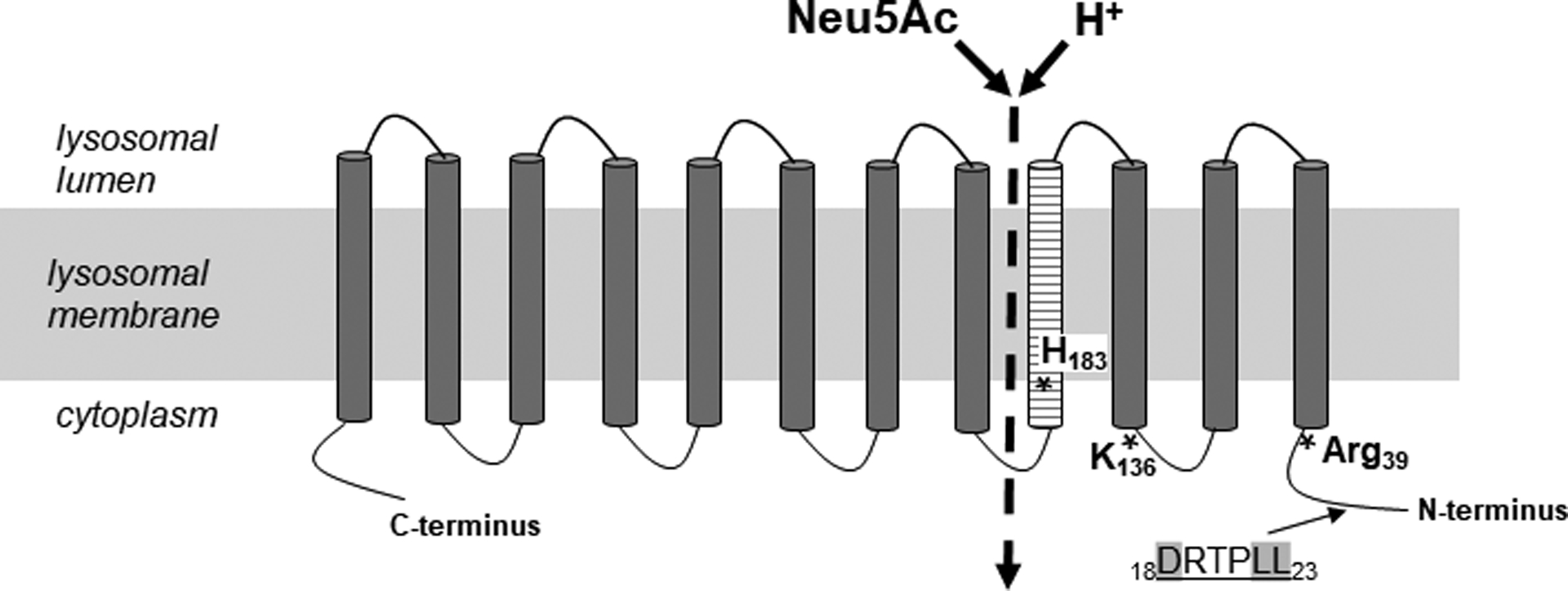
Lysosomal free sialic acid storage disorder (FSASD) is an extremely rare, autosomal recessive, neurodegenerative, multisystemic disorder caused by defects in the lysosomal sialic acid membrane exporter SLC17A5 (sialin). SLC17A5 defects cause free sialic acid and some other acidic hexoses to accumulate in lysosomes, resulting in enlarged lysosomes in some cell types and 10-100-fold increased urinary excretion of free sialic acid. Clinical features of FSASD include coarse facial features, organomegaly, and progressive neurodegenerative symptoms with cognitive impairment, cerebellar ataxia and muscular hypotonia. Central hypomyelination with cerebellar atrophy and thinning of the corpus callosum are also prominent disease features. Around 200 FSASD cases are reported worldwide, with the clinical spectrum ranging from a severe infantile onset form, often lethal in early childhood, to a mild, less severe form with subjects living into adulthood, also called Salla disease. The pathobiology of FSASD remains poorly understood and FSASD is likely underdiagnosed. Known patients have experienced a diagnostic delay due to the rarity of the disorder, absence of routine urine sialic acid testing, and non-specific clinical symptoms, including developmental delay, ataxia and infantile hypomyelination. There is no approved therapy for FSASD. We initiated a multidisciplinary collaborative effort involving worldwide academic clinical and scientific FSASD experts, the National Institutes of Health (USA), and the FSASD patient advocacy group (Salla Treatment and Research [S.T.A.R.] Foundation) to overcome the scientific, clinical and financial challenges facing the development of new treatments for FSASD. We aim to collect data that incentivize industry to further develop, obtain approval for, and commercialize FSASD treatments. This review summarizes current aspects of FSASD diagnosis, prevalence, etiology, and disease models, as well as challenges on the path to therapeutic approaches for FSASD.
Keywords: Hypomyelination; Infantile sialic acid storage disorder; Lysosomal membrane transporter; N-acetylneuraminic acid; SLC17A5; Salla disease; Sialic acid.
Copyright © 2021. Published by Elsevier B.V.
Conflict of interest statement
Figures
Similar articles
-
Generation and characterization of two iPSC lines derived from subjects with Free Sialic Acid Storage Disorder (FSASD).Stem Cell Res. 2024 Dec;81:103600. doi: 10.1016/j.scr.2024.103600. Epub 2024 Oct 22. Stem Cell Res. 2024. PMID: 39461116
-
Identification of a large intronic transposal insertion in SLC17A5 causing sialic acid storage disease.Orphanet J Rare Dis. 2017 Feb 10;12(1):28. doi: 10.1186/s13023-017-0584-6. Orphanet J Rare Dis. 2017. PMID: 28187749 Free PMC article.
-
Clinical, biochemical, and molecular diagnosis of a free sialic acid storage disease patient of moderate severity.Mol Genet Metab. 2004 Jun;82(2):137-43. doi: 10.1016/j.ymgme.2004.03.001. Mol Genet Metab. 2004. PMID: 15172001
-
A New Patient With Intermediate Severe Salla Disease With Hypomyelination: A Literature Review for Salla Disease.Pediatr Neurol. 2017 Sep;74:87-91.e2. doi: 10.1016/j.pediatrneurol.2017.05.022. Epub 2017 Jun 1. Pediatr Neurol. 2017. PMID: 28662915 Review.
-
Sialic acid storage disease and related disorders.Genet Test. 2003 Summer;7(2):113-21. doi: 10.1089/109065703322146795. Genet Test. 2003. PMID: 12885332 Review.
Cited by
-
Soft X-ray spectromicroscopy of human fibroblasts with impaired sialin function.RSC Adv. 2024 Sep 10;14(39):28797-28806. doi: 10.1039/d4ra05520a. eCollection 2024 Sep 4. RSC Adv. 2024. PMID: 39257666 Free PMC article.
-
Psychiatric symptoms in Salla disease.Eur Child Adolesc Psychiatry. 2023 Oct;32(10):2043-2047. doi: 10.1007/s00787-022-02031-5. Epub 2022 Jul 7. Eur Child Adolesc Psychiatry. 2023. PMID: 35796883 Free PMC article. Review.
-
Base editing corrects the common Salla disease SLC17A5 c.115C>T variant.Mol Ther Nucleic Acids. 2023 Aug 26;34:102022. doi: 10.1016/j.omtn.2023.08.024. eCollection 2023 Dec 12. Mol Ther Nucleic Acids. 2023. PMID: 37727271 Free PMC article.
-
Using team-based precision medicine to advance understanding of rare genetic brain disorders.J Neurodev Disord. 2024 Mar 15;16(1):10. doi: 10.1186/s11689-024-09518-z. J Neurodev Disord. 2024. PMID: 38491427 Free PMC article. Review.
-
Assessment of genes involved in lysosomal diseases using the ClinGen clinical validity framework.Mol Genet Metab. 2024 Nov;143(3):108593. doi: 10.1016/j.ymgme.2024.108593. Epub 2024 Oct 12. Mol Genet Metab. 2024. PMID: 39426251
References
-
- Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, van der Spek PJ, Mancini GM, A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases, Nat Genet 23 (1999) 462–465. - PubMed
-
- Adams D, Wasserstein M, Free Sialic Acid Storage Disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews, University of Washington, Seattle (WA), 2003. [Updated 2020], p. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1470/. - PubMed
-
- Aula P, Gahl WA, Disorders of Free Sialic Acid Storage. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, G.A. M (Eds.), The Online Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, 2019, p. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225891389.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
