

Transcriptional activation of antioxidant gene expression by Nrf2 protects against mitochondrial dysfunction and neuronal death associated with acute and chronic neurodegeneration
- PMID: 32061629
- PMCID: PMC8627637
- DOI: 10.1016/j.expneurol.2020.113247
Transcriptional activation of antioxidant gene expression by Nrf2 protects against mitochondrial dysfunction and neuronal death associated with acute and chronic neurodegeneration
Abstract
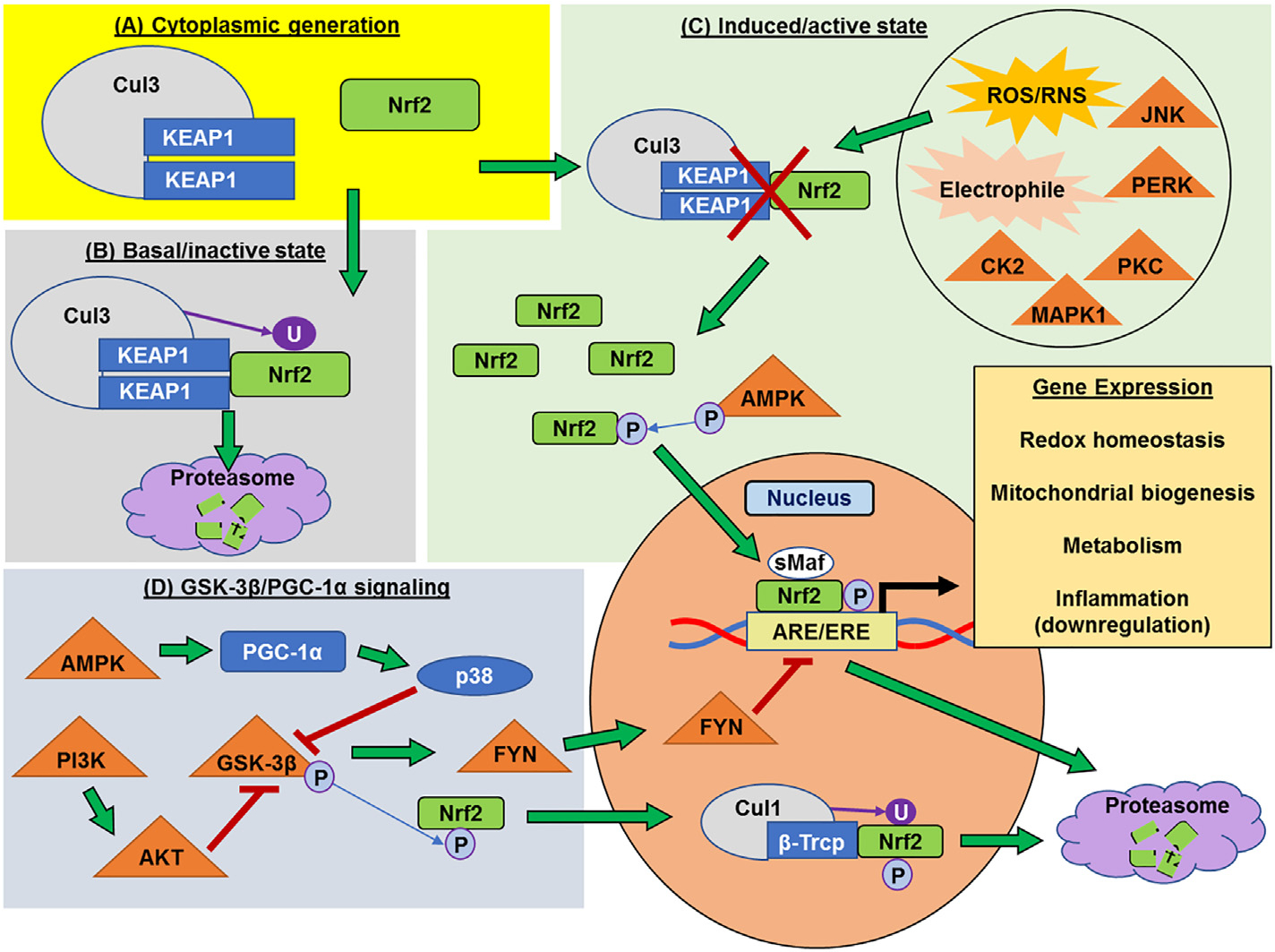
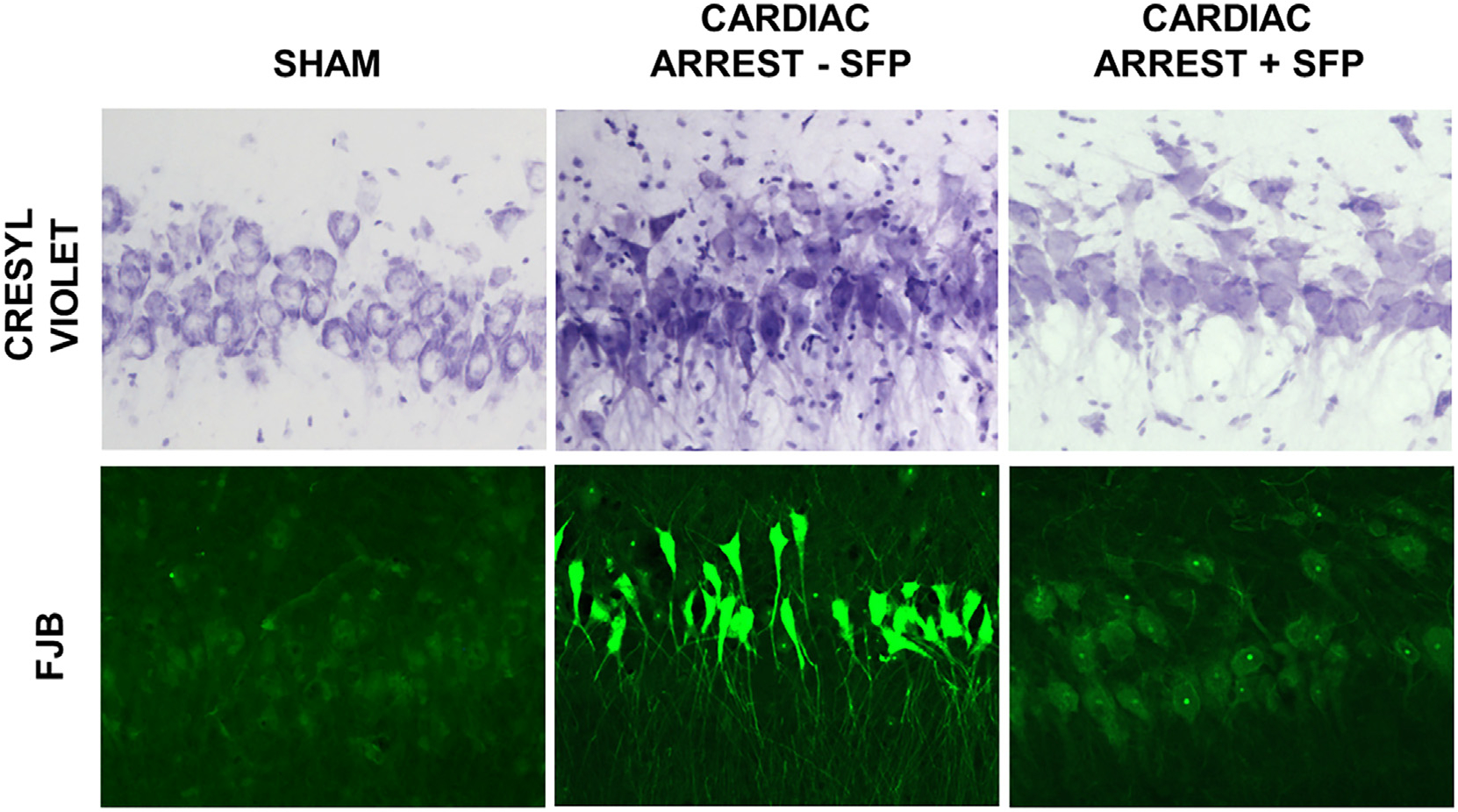
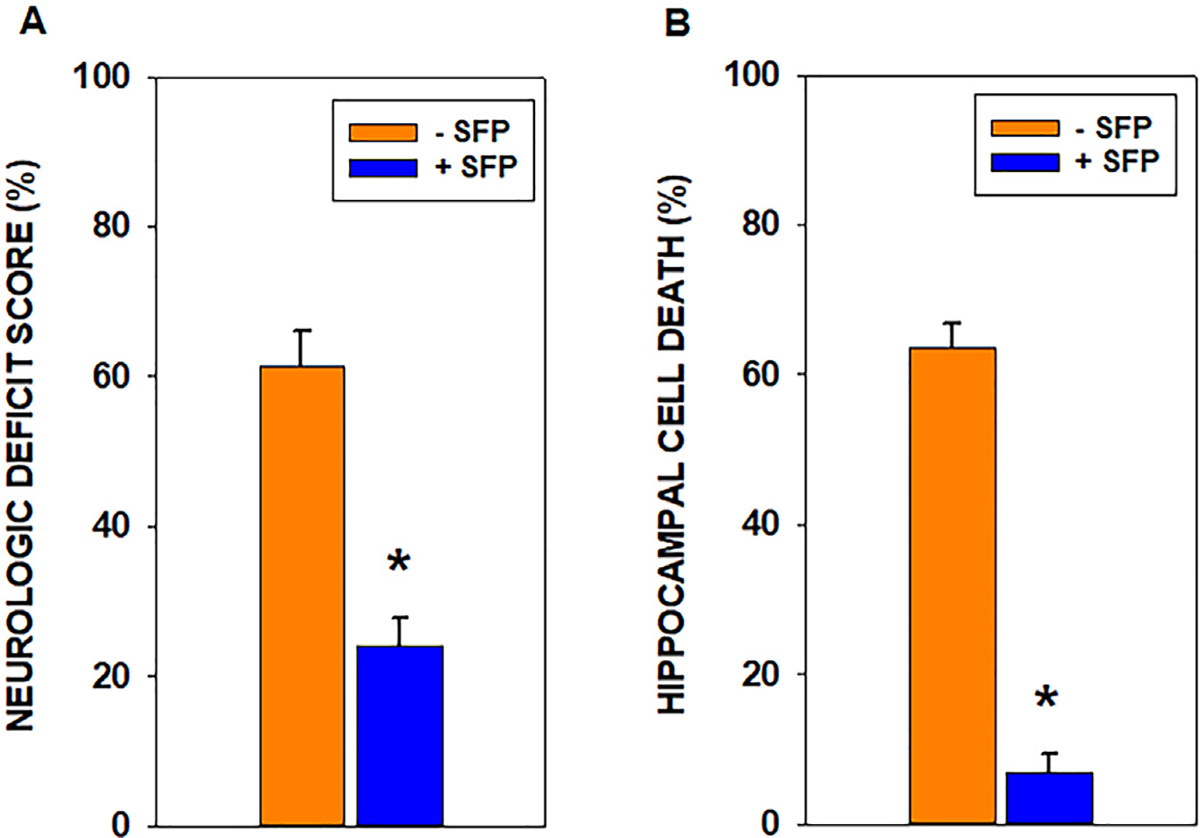
Mitochondria are both a primary source of reactive oxygen species (ROS) and a sensitive target of oxidative stress; damage to mitochondria can result in bioenergetic dysfunction and both necrotic and apoptotic cell death. These relationships between mitochondria and cell death are particularly strong in both acute and chronic neurodegenerative disorders. ROS levels are affected by both the production of superoxide and its toxic metabolites and by antioxidant defense mechanisms. Mitochondrial antioxidant activities include superoxide dismutase 2, glutathione peroxidase and reductase, and intramitochondrial glutathione. When intracellular conditions disrupt the homeostatic balance between ROS production and detoxification, a net increase in ROS and an oxidized shift in cellular redox state ensues. Cells respond to this imbalance by increasing the expression of genes that code for proteins that protect against oxidative stress and inhibit cytotoxic oxidation of proteins, DNA, and lipids. If, however, the genomic response to mitochondrial oxidative stress is insufficient to maintain homeostasis, mitochondrial bioenergetic dysfunction and release of pro-apoptotic mitochondrial proteins into the cytosol initiate a variety of cell death pathways, ultimately resulting in potentially lethal damage to vital organs, including the brain. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a translational activating protein that enters the nucleus in response to oxidative stress, resulting in increased expression of numerous cytoprotective genes, including genes coding for mitochondrial and non-mitochondrial antioxidant proteins. Many experimental and some FDA-approved drugs promote this process. Since mitochondria are targets of ROS, it follows that protection against mitochondrial oxidative stress by the Nrf2 pathway of gene expression contributes to neuroprotection by these drugs. This document reviews the evidence that Nrf2 activation increases mitochondrial antioxidants, thereby protecting mitochondria from dysfunction and protecting neural cells from damage and death. New experimental results are provided demonstrating that post-ischemic administration of the Nrf2 activator sulforaphane protects against hippocampal neuronal death and neurologic injury in a clinically-relevant animal model of cardiac arrest and resuscitation.
Keywords: Calcium; Excitotoxicity; Inflammation; Oxidative stress; Reactive nitrogen species; Reactive oxygen species; Sulforaphane.
Copyright © 2020. Published by Elsevier Inc.
Conflict of interest statement
Declaration of Competing Interest The authors have nothing to declare.
Figures
Similar articles
-
Acute exercise stress promotes Ref1/Nrf2 signalling and increases mitochondrial antioxidant activity in skeletal muscle.Exp Physiol. 2016 Mar;101(3):410-20. doi: 10.1113/EP085493. Epub 2016 Jan 23. Exp Physiol. 2016. PMID: 26682532
-
Neuroprotection through stimulation of mitochondrial antioxidant protein expression.J Alzheimers Dis. 2010;20 Suppl 2:S427-37. doi: 10.3233/JAD-2010-100519. J Alzheimers Dis. 2010. PMID: 20463397 Review.
-
Activation of the Nrf2-regulated antioxidant cell response inhibits HEMA-induced oxidative stress and supports cell viability.Biomaterials. 2015 Jul;56:114-28. doi: 10.1016/j.biomaterials.2015.03.047. Epub 2015 Apr 16. Biomaterials. 2015. PMID: 25934285
-
Homoeriodictyol protects human endothelial cells against oxidative insults through activation of Nrf2 and inhibition of mitochondrial dysfunction.Vascul Pharmacol. 2018 Oct;109:72-82. doi: 10.1016/j.vph.2018.06.007. Epub 2018 Jun 11. Vascul Pharmacol. 2018. PMID: 29902531
-
Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds.Pharmacol Res. 2016 Jan;103:80-94. doi: 10.1016/j.phrs.2015.11.019. Epub 2015 Nov 25. Pharmacol Res. 2016. PMID: 26626189 Review.
Cited by
-
Beta-Hydroxybutyrate Mitigates Sensorimotor and Cognitive Impairments in a Photothrombosis-Induced Ischemic Stroke in Mice.Int J Mol Sci. 2024 May 24;25(11):5710. doi: 10.3390/ijms25115710. Int J Mol Sci. 2024. PMID: 38891898 Free PMC article.
-
Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases?Antioxidants (Basel). 2022 Jan 15;11(1):165. doi: 10.3390/antiox11010165. Antioxidants (Basel). 2022. PMID: 35052668 Free PMC article. Review.
-
Calpain inhibitor MDL28170 alleviates cerebral ischemia‑reperfusion injury by suppressing inflammation and autophagy in a rat model of cardiac arrest.Exp Ther Med. 2023 Mar 17;25(5):196. doi: 10.3892/etm.2023.11895. eCollection 2023 May. Exp Ther Med. 2023. PMID: 37090078 Free PMC article.
-
Evaluation of arsenic metabolism and tight junction injury after exposure to arsenite and monomethylarsonous acid using a rat in vitro blood-Brain barrier model.PLoS One. 2023 Nov 30;18(11):e0295154. doi: 10.1371/journal.pone.0295154. eCollection 2023. PLoS One. 2023. PMID: 38032905 Free PMC article.
-
Common Protective Strategies in Neurodegenerative Disease: Focusing on Risk Factors to Target the Cellular Redox System.Oxid Med Cell Longev. 2020 Aug 1;2020:8363245. doi: 10.1155/2020/8363245. eCollection 2020. Oxid Med Cell Longev. 2020. PMID: 32832006 Free PMC article. Review.
References
-
- Abdullah A, Kitteringham NR, Jenkins RE, Goldring C, Higgins L, Yamamoto M, Hayes J, Park BK, 2012. Analysis of the role of Nrf2 in the expression of liver proteins in mice using two-dimensional gel-based proteomics. Pharmacol. Rep. 64, 680–697. - PubMed
-
- Acaz-Fonseca E, Duran JC, Carrero P, Garcia-Segura LM, Arevalo MA, 2015. Sex differences in glia reactivity after cortical brain injury. Glia 63, 1966–1981. - PubMed
-
- Ahmed SMU, Luo L, Namani A, Wang XJ, Tang X, 2017. Nrf2 signaling pathway: pivotal roles in inflammation. BBA-Mol Basis Dis 1863, 585–597. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
