

De novo substitutions of TRPM3 cause intellectual disability and epilepsy
- PMID: 31278393
- PMCID: PMC6777445
- DOI: 10.1038/s41431-019-0462-x
De novo substitutions of TRPM3 cause intellectual disability and epilepsy
Abstract
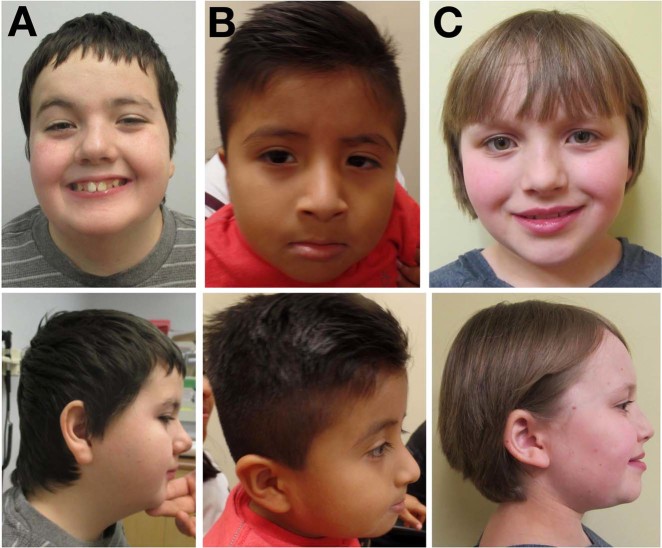
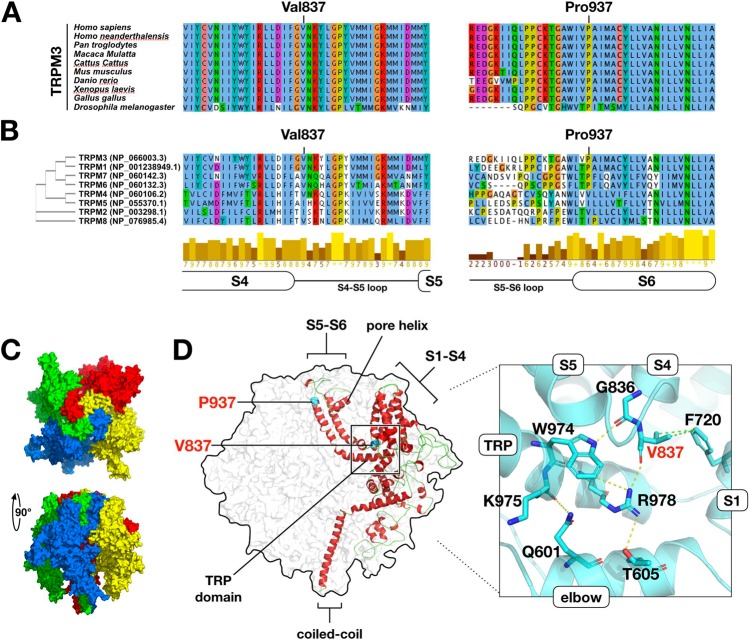
The developmental and epileptic encephalopathies (DEE) are a heterogeneous group of chronic encephalopathies frequently associated with rare de novo nonsynonymous coding variants in neuronally expressed genes. Here, we describe eight probands with a DEE phenotype comprising intellectual disability, epilepsy, and hypotonia. Exome trio analysis showed de novo variants in TRPM3, encoding a brain-expressed transient receptor potential channel, in each. Seven probands were identically heterozygous for a recurrent substitution, p.(Val837Met), in TRPM3's S4-S5 linker region, a conserved domain proposed to undergo conformational change during gated channel opening. The eighth individual was heterozygous for a proline substitution, p.(Pro937Gln), at the boundary between TRPM3's flexible pore-forming loop and an adjacent alpha-helix. General-population truncating variants and microdeletions occur throughout TRPM3, suggesting a pathomechanism other than simple haploinsufficiency. We conclude that de novo variants in TRPM3 are a cause of intellectual disability and epilepsy.
Conflict of interest statement
KMW is an employee of GeneDx, Inc. The other authors declare that they have no conflict of interest.
Figures
Similar articles
-
A Chinese patient with developmental and epileptic encephalopathies (DEE) carrying a TRPM3 gene mutation: a paediatric case report.BMC Pediatr. 2021 Jun 1;21(1):256. doi: 10.1186/s12887-021-02719-8. BMC Pediatr. 2021. PMID: 34074259 Free PMC article.
-
Confirmation and Expansion of the Phenotype Associated with the Recurrent p.Val837Met Variant in TRPM3.Eur J Med Genet. 2020 Aug;63(8):103942. doi: 10.1016/j.ejmg.2020.103942. Epub 2020 May 18. Eur J Med Genet. 2020. PMID: 32439617
-
Gain of channel function and modified gating properties in TRPM3 mutants causing intellectual disability and epilepsy.Elife. 2020 May 19;9:e57190. doi: 10.7554/eLife.57190. Elife. 2020. PMID: 32427099 Free PMC article.
-
Neurodevelopmental disorders caused by variants in TRPM3.Biochim Biophys Acta Mol Cell Res. 2024 Jun;1871(5):119709. doi: 10.1016/j.bbamcr.2024.119709. Epub 2024 Mar 23. Biochim Biophys Acta Mol Cell Res. 2024. PMID: 38522727 Review.
-
The newest TRP channelopathy: Gain of function TRPM3 mutations cause epilepsy and intellectual disability.Channels (Austin). 2021 Dec;15(1):386-397. doi: 10.1080/19336950.2021.1908781. Channels (Austin). 2021. PMID: 33853504 Free PMC article. Review.
Cited by
-
TRPM3 Channels Play Roles in Heat Hypersensitivity and Spontaneous Pain after Nerve Injury.J Neurosci. 2021 Mar 17;41(11):2457-2474. doi: 10.1523/JNEUROSCI.1551-20.2020. Epub 2021 Jan 21. J Neurosci. 2021. PMID: 33478988 Free PMC article.
-
Functional expression and pharmacological modulation of TRPM3 in human sensory neurons.Br J Pharmacol. 2020 Jun;177(12):2683-2695. doi: 10.1111/bph.14994. Epub 2020 Mar 5. Br J Pharmacol. 2020. PMID: 31985045 Free PMC article.
-
The Underlying Mechanism of Modulation of Transient Receptor Potential Melastatin 3 by protons.Front Pharmacol. 2021 Feb 2;12:632711. doi: 10.3389/fphar.2021.632711. eCollection 2021. Front Pharmacol. 2021. PMID: 33603674 Free PMC article.
-
Novel Genetic Diagnoses in Septo-Optic Dysplasia.Genes (Basel). 2022 Jun 28;13(7):1165. doi: 10.3390/genes13071165. Genes (Basel). 2022. PMID: 35885948 Free PMC article.
-
TRP channels in health and disease at a glance.J Cell Sci. 2021 Jul 1;134(13):jcs258372. doi: 10.1242/jcs.258372. Epub 2021 Jul 13. J Cell Sci. 2021. PMID: 34254641 Free PMC article.
