

Pathophysiology of pachyonychia congenita-associated palmoplantar keratoderma: new insights into skin epithelial homeostasis and avenues for treatment
- PMID: 31021398
- PMCID: PMC6814456
- DOI: 10.1111/bjd.18033
Pathophysiology of pachyonychia congenita-associated palmoplantar keratoderma: new insights into skin epithelial homeostasis and avenues for treatment
Abstract
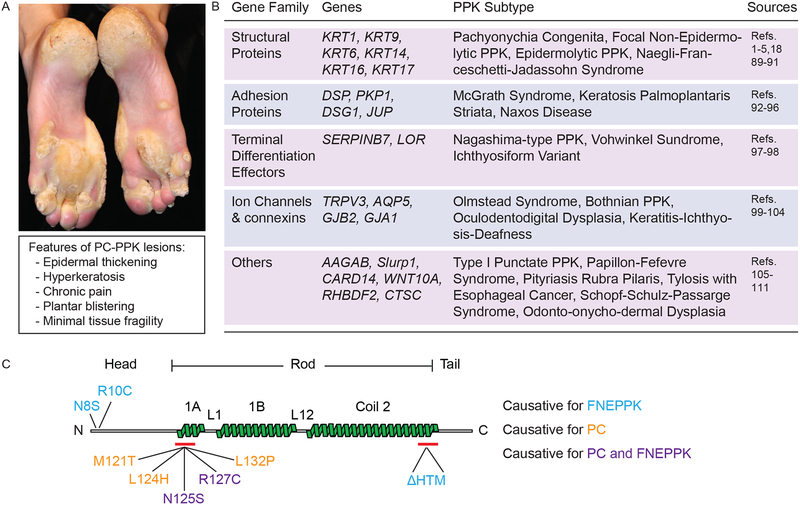
Background: Pachyonychia congenita (PC), a rare genodermatosis, primarily affects ectoderm-derived epithelial appendages and typically includes oral leukokeratosis, nail dystrophy and very painful palmoplantar keratoderma (PPK). PC dramatically impacts quality of life although it does not affect lifespan. PC can arise from mutations in any of the wound-repair-associated keratin genes KRT6A, KRT6B, KRT6C, KRT16 or KRT17. There is no cure for this condition, and current treatment options for PC symptoms are limited and palliative in nature.
Objectives: This review focuses on recent progress made towards understanding the pathophysiology of PPK lesions, the most prevalent and debilitating of all PC symptoms.
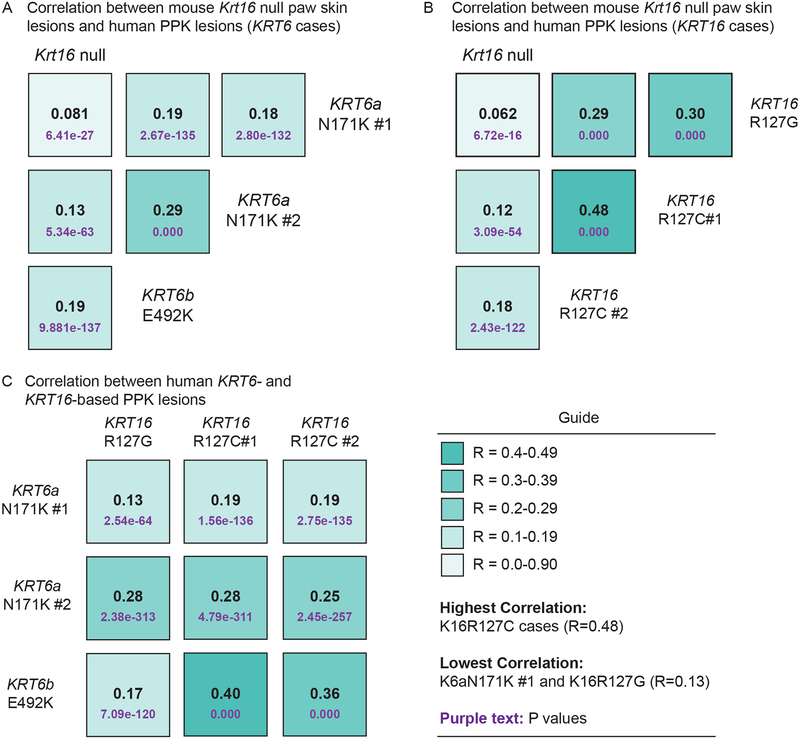
Methods: We reviewed the relevant literature with a particular focus on the Krt16 null mouse, which spontaneously develops footpad lesions that mimic several aspects of PC-associated PPK.
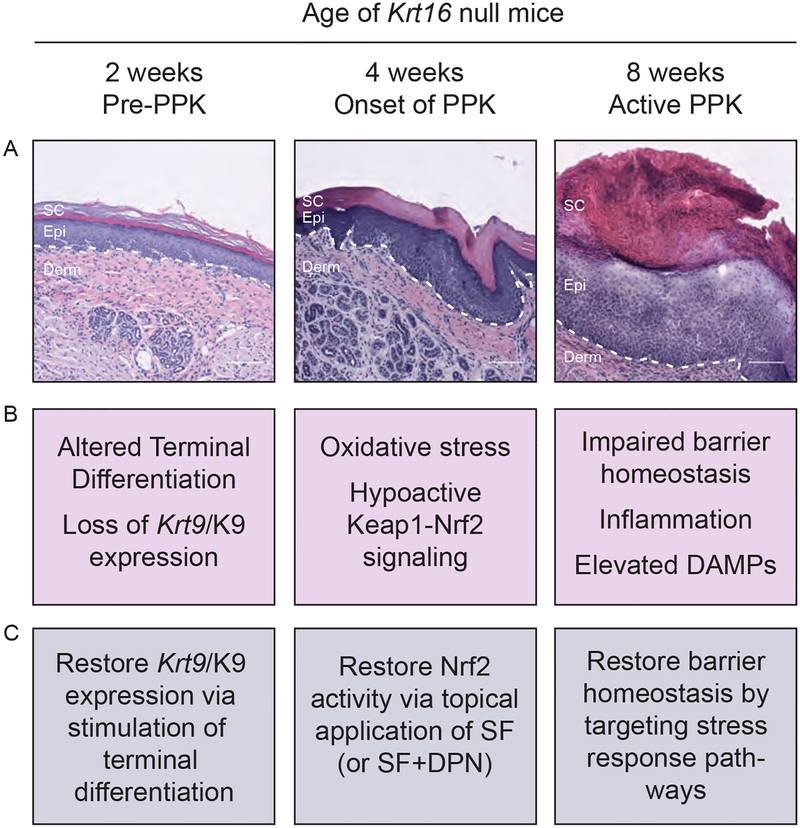
Results: There are three main stages of progression of PPK-like lesions in Krt16 null mice. Ahead of lesion onset, keratinocytes in the palmoplantar (footpad) skin exhibit specific defects in terminal differentiation, including loss of Krt9 expression. At the time of PPK onset, there is elevated oxidative stress and hypoactive Keap1-Nrf2 signalling. During active PPK, there is a profound defect in the ability of the epidermis to maintain or return to normal homeostasis.
Conclusions: The progress made suggests new avenues to explore for the treatment of PC-based PPK and deepens our understanding of the mechanisms controlling skin tissue homeostasis. What's already known about this topic? Pachyonychia congenita (PC) is a rare genodermatosis caused by mutations in KRT6A, KRT6B, KRT6C, KRT16 and KRT17, which are normally expressed in skin appendages and induced following injury. Individuals with PC present with multiple clinical symptoms that usually include thickened and dystrophic nails, palmoplantar keratoderma (PPK), glandular cysts and oral leukokeratosis. The study of PC pathophysiology is made challenging because of its low incidence and high complexity. There is no cure or effective treatment for PC. What does this study add? This text reviews recent progress made when studying the pathophysiology of PPK associated with PC. This recent progress points to new possibilities for devising effective therapeutics that may complement current palliative strategies.
© 2019 British Association of Dermatologists.
Conflict of interest statement
Figures
Comment in
-
Current mysteries of pachyonychia congenita.Br J Dermatol. 2020 Mar;182(3):525-526. doi: 10.1111/bjd.18688. Epub 2019 Dec 8. Br J Dermatol. 2020. PMID: 31814103 No abstract available.
Similar articles
-
Altered keratinocyte differentiation is an early driver of keratin mutation-based palmoplantar keratoderma.Hum Mol Genet. 2019 Jul 1;28(13):2255-2270. doi: 10.1093/hmg/ddz050. Hum Mol Genet. 2019. PMID: 31220272 Free PMC article.
-
Oxidative stress and dysfunctional NRF2 underlie pachyonychia congenita phenotypes.J Clin Invest. 2016 Jun 1;126(6):2356-66. doi: 10.1172/JCI84870. Epub 2016 May 16. J Clin Invest. 2016. PMID: 27183391 Free PMC article.
-
Keratin 16-null mice develop palmoplantar keratoderma, a hallmark feature of pachyonychia congenita and related disorders.J Invest Dermatol. 2012 May;132(5):1384-91. doi: 10.1038/jid.2012.6. Epub 2012 Feb 16. J Invest Dermatol. 2012. PMID: 22336941 Free PMC article.
-
A KRT16 mutation in the first Chinese pedigree with Pachyonychia congenita and review of the literatures.J Cosmet Dermatol. 2019 Dec;18(6):1930-1934. doi: 10.1111/jocd.12905. Epub 2019 Mar 12. J Cosmet Dermatol. 2019. PMID: 30859684 Review.
-
Pachyonychia Congenita: A Spectrum of KRT6a Mutations in Australian Patients.Pediatr Dermatol. 2016 May;33(3):337-42. doi: 10.1111/pde.12841. Epub 2016 Apr 4. Pediatr Dermatol. 2016. PMID: 27041546 Review.
Cited by
-
Distinctions in the Management, Patient Impact, and Clinical Profiles of Pachyonychia Congenita Subtypes.Skin Appendage Disord. 2021 Apr;7(3):194-202. doi: 10.1159/000513340. Epub 2021 Feb 5. Skin Appendage Disord. 2021. PMID: 34055907 Free PMC article.
-
Pachyonychia Congenita.Indian J Pediatr. 2024 Mar;91(3):300-301. doi: 10.1007/s12098-023-04567-z. Epub 2023 Jun 8. Indian J Pediatr. 2024. PMID: 37289311 No abstract available.
-
Inhibiting EGFR Signaling Holds Promise for Treating Palmoplantar Keratodermas.J Invest Dermatol. 2023 Feb;143(2):185-188. doi: 10.1016/j.jid.2022.09.653. J Invest Dermatol. 2023. PMID: 36681421 Free PMC article. No abstract available.
-
Repeated stress to the skin amplifies neutrophil infiltration in a keratin 17- and PKCα-dependent manner.PLoS Biol. 2024 Aug 19;22(8):e3002779. doi: 10.1371/journal.pbio.3002779. eCollection 2024 Aug. PLoS Biol. 2024. PMID: 39159283 Free PMC article.
-
The Treatment of Refractory Vitiligo With Autologous Cultured Epithelium Grafting: A Real-World Retrospective Cohort Study.Stem Cells Transl Med. 2024 May 14;13(5):415-424. doi: 10.1093/stcltm/szae009. Stem Cells Transl Med. 2024. PMID: 38513284 Free PMC article.
References
-
- Leachman SA, Kaspar RL, Fleckman P et al. Clinical and pathological features of pachyonychia congenita. The journal of investigative dermatology. Symposium proceedings 2005; 10: 3–17. - PubMed
-
- Wilson NJ, Messenger AG, Leachman SA et al. Keratin K6c mutations cause focal palmoplantar keratoderma. The Journal of investigative dermatology 2010; 130: 425–9. - PubMed
-
- McLean WH, Rugg EL, Lunny DP et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nature genetics 1995; 9: 273–8. - PubMed
-
- Smith FJ, Jonkman MF, van Goor H et al. A mutation in human keratin K6b produces a phenocopy of the K17 disorder pachyonychia congenita type 2. Human molecular genetics 1998; 7: 1143–8. - PubMed
-
- Bowden PE, Haley JL, Kansky A et al. Mutation of a type II keratin gene (K6a) in pachyonychia congenita. Nature genetics 1995; 10: 363–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
