

Mitochondrial Neurogastrointestinal Encephalomyopathy: Into the Fourth Decade, What We Have Learned So Far
- PMID: 30627136
- PMCID: PMC6309918
- DOI: 10.3389/fgene.2018.00669
Mitochondrial Neurogastrointestinal Encephalomyopathy: Into the Fourth Decade, What We Have Learned So Far
Abstract
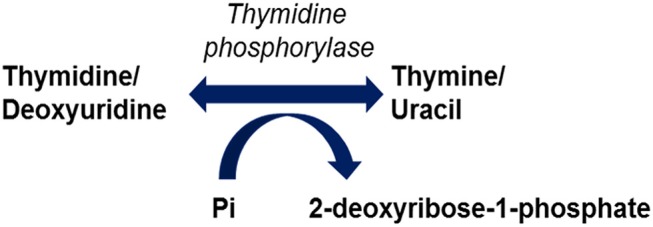
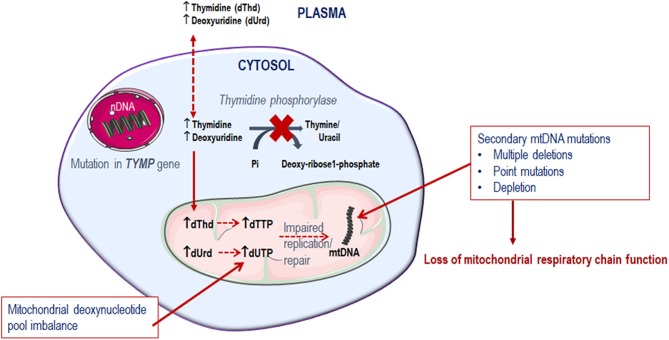
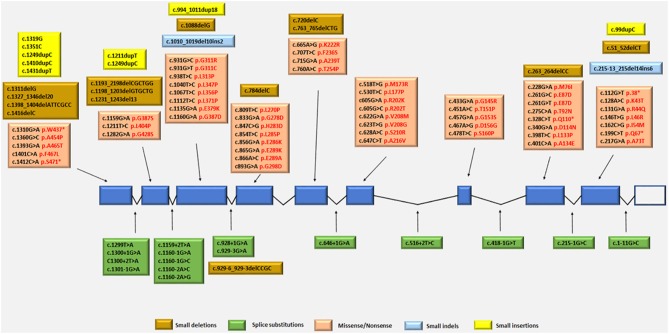
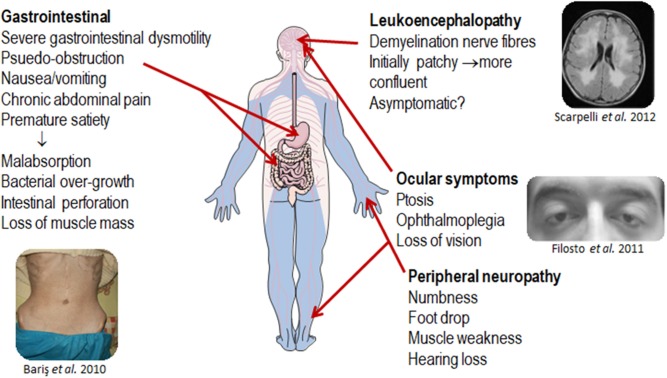
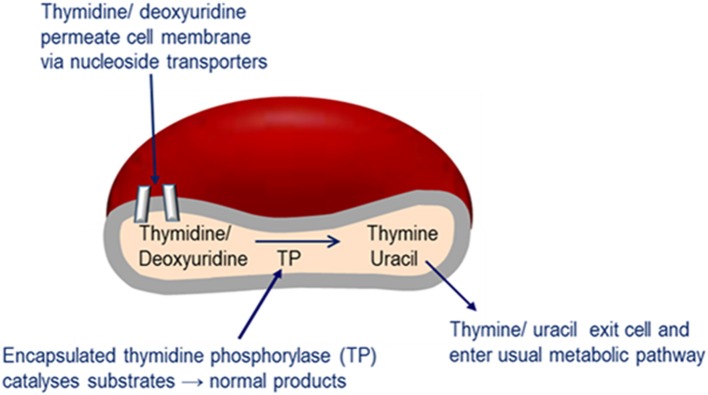
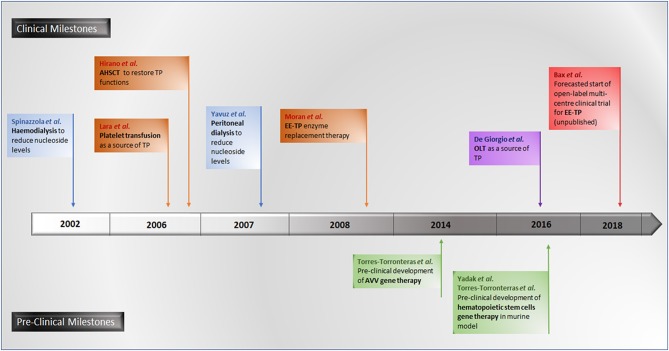
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an ultra-rare metabolic autosomal recessive disease, caused by mutations in the nuclear gene TYMP which encodes the enzyme thymidine phosphorylase. The resulting enzyme deficiency leads to a systemic accumulation of the deoxyribonucleosides thymidine and deoxyuridine, and ultimately mitochondrial failure due to a progressive acquisition of secondary mitochondrial DNA (mtDNA) mutations and mtDNA depletion. Clinically, MNGIE is characterized by gastrointestinal and neurological manifestations, including cachexia, gastrointestinal dysmotility, peripheral neuropathy, leukoencephalopathy, ophthalmoplegia and ptosis. The disease is progressively degenerative and leads to death at an average age of 37.6 years. As with the vast majority of rare diseases, patients with MNGIE face a number of unmet needs related to diagnostic delays, a lack of approved therapies, and non-specific clinical management. We provide here a comprehensive collation of the available knowledge of MNGIE since the disease was first described 42 years ago. This review includes symptomatology, diagnostic procedures and hurdles, in vitro and in vivo disease models that have enhanced our understanding of the disease pathology, and finally experimental therapeutic approaches under development. The ultimate aim of this review is to increase clinical awareness of MNGIE, thereby reducing diagnostic delay and improving patient access to putative treatments under investigation.
Keywords: MNGIE; TYMP; deoxyribonucleoside; mitochondrial DNA; mitochondrial disease; mitochondrial neurogastrointestinal encephalomyopathy; rare disease; thymidine phosphorylase.
Figures

Similar articles
-
Mitochondrial neurogastrointestinal encephalomyopathy: approaches to diagnosis and treatment.J Transl Genet Genom. 2020 Mar 30;4:1-16. doi: 10.20517/jtgg.2020.08. J Transl Genet Genom. 2020. PMID: 32914088 Free PMC article.
-
Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1).J Clin Med. 2018 Oct 26;7(11):389. doi: 10.3390/jcm7110389. J Clin Med. 2018. PMID: 30373120 Free PMC article. Review.
-
Mitochondrial Neurogastrointestinal Encephalomyopathy Disease in Three Siblings from Pakistan with a Novel Mutation.J Pediatr Genet. 2019 Mar;8(1):15-19. doi: 10.1055/s-0038-1661411. Epub 2018 Jun 30. J Pediatr Genet. 2019. PMID: 30775048 Free PMC article.
-
Mitochondrial Neurogastrointestinal Encephalomyopathy Caused by Thymidine Phosphorylase Enzyme Deficiency: From Pathogenesis to Emerging Therapeutic Options.Front Cell Neurosci. 2017 Feb 15;11:31. doi: 10.3389/fncel.2017.00031. eCollection 2017. Front Cell Neurosci. 2017. PMID: 28261062 Free PMC article. Review.
-
Meningoencephalitis in a novel mutation in MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) ending a familial diagnostic odyssey: A case series report.J Cent Nerv Syst Dis. 2024 Mar 27;16:11795735241241423. doi: 10.1177/11795735241241423. eCollection 2024. J Cent Nerv Syst Dis. 2024. PMID: 38550250 Free PMC article.
Cited by
-
Mitochondrial Epilepsy, a Challenge for Neurologists.Int J Mol Sci. 2022 Oct 30;23(21):13216. doi: 10.3390/ijms232113216. Int J Mol Sci. 2022. PMID: 36362003 Free PMC article. Review.
-
COQ7 defect causes prenatal onset of mitochondrial CoQ10 deficiency with cardiomyopathy and gastrointestinal obstruction.Eur J Hum Genet. 2024 Aug;32(8):938-946. doi: 10.1038/s41431-024-01615-w. Epub 2024 May 3. Eur J Hum Genet. 2024. PMID: 38702428 Free PMC article.
-
Successful Sequential Liver and Isolated Intestine Transplantation for Mitochondrial Neurogastrointestinal Encephalopathy Syndrome: A Case Report.Ann Transplant. 2024 Feb 27;29:e941881. doi: 10.12659/AOT.941881. Ann Transplant. 2024. PMID: 38409779 Free PMC article.
-
Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in Mitochondrial Neurogastrointestinal Encephalomyopathy.J Clin Med. 2019 Apr 5;8(4):457. doi: 10.3390/jcm8040457. J Clin Med. 2019. PMID: 30959750 Free PMC article.
-
Ongoing Developments and Clinical Progress in Drug-Loaded Red Blood Cell Technologies.BioDrugs. 2020 Jun;34(3):265-272. doi: 10.1007/s40259-020-00415-0. BioDrugs. 2020. PMID: 32198632 Free PMC article. Review.
References
-
- Aksoy F., Demirel G., Bilgiç T., Güngör I. G., Ozçelik A. (2005). A previously diagnosed mitochondrial neurogastrointestinal encephalomyopathy patient presenting with perforated ileal diverticulitis. Turk. J. Gastroenterol. 16, 228–231. - PubMed
-
- Anuras S., Mitros F. A., Nowak T. V., Ionasescu V. V., Gurll N. J., Christensen J., et al. . (1983). A familial visceral myopathy with external ophthalmoplegia and autosomal recessive transmission. Gastroenterology 84, 346–353. - PubMed
-
- Asai K., Nakanishi K., Isobe I., Eksioglu Y. Z., Hirano A., Hama K., et al. . (1992b). Neurotrophic action of gliostatin on cortical neurons. Identity of gliostatin and platelet-derived endothelial cell growth factor J. Biol. Chem. 267, 20311–20316. - PubMed
-
- Baker M. K., Schutte C. M., Ranchhod N., Brittain D., van Rensburg J. E. (2017). Transient clinical improvement of a mitochondrial neurogastrointestinal encephalomyopathy-like syndrome after allogeneic haematopoietic stem cell transplantation. BMJ Case Rep. 2017:bcr-2016-218276. 10.1136/bcr-2016-218276 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials