

Short stature and hypoparathyroidism in a child with Kenny-Caffey syndrome type 2 due to a novel mutation in FAM111A gene
- PMID: 28138333
- PMCID: PMC5264330
- DOI: 10.1186/s13633-016-0041-7
Short stature and hypoparathyroidism in a child with Kenny-Caffey syndrome type 2 due to a novel mutation in FAM111A gene
Abstract
Background: Hypoparathyroidism in children is a heterogeneous group with diverse genetic etiologies. To aid clinicians in the investigation and management of children with hypoparathyroidism, we describe the phenotype of a 6-year-old child with hypoparathyroidism and short stature diagnosed with Kenny-Caffey syndrome (KCS) Type 2 and the subsequent response to growth hormone (GH) treatment.
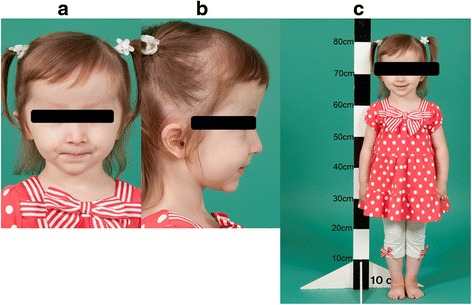
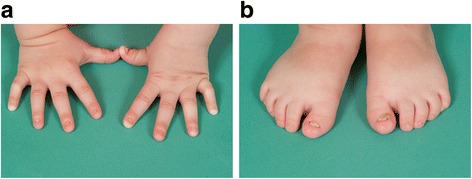
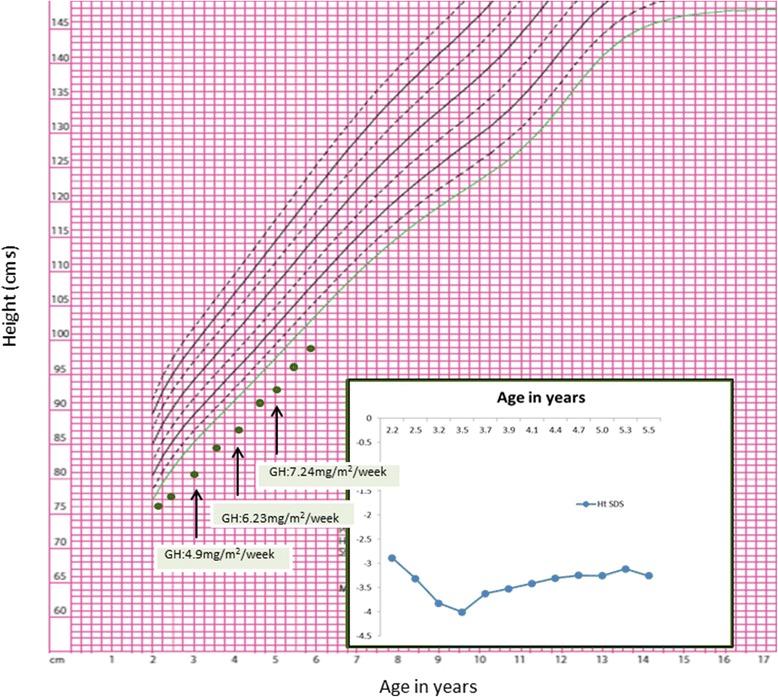
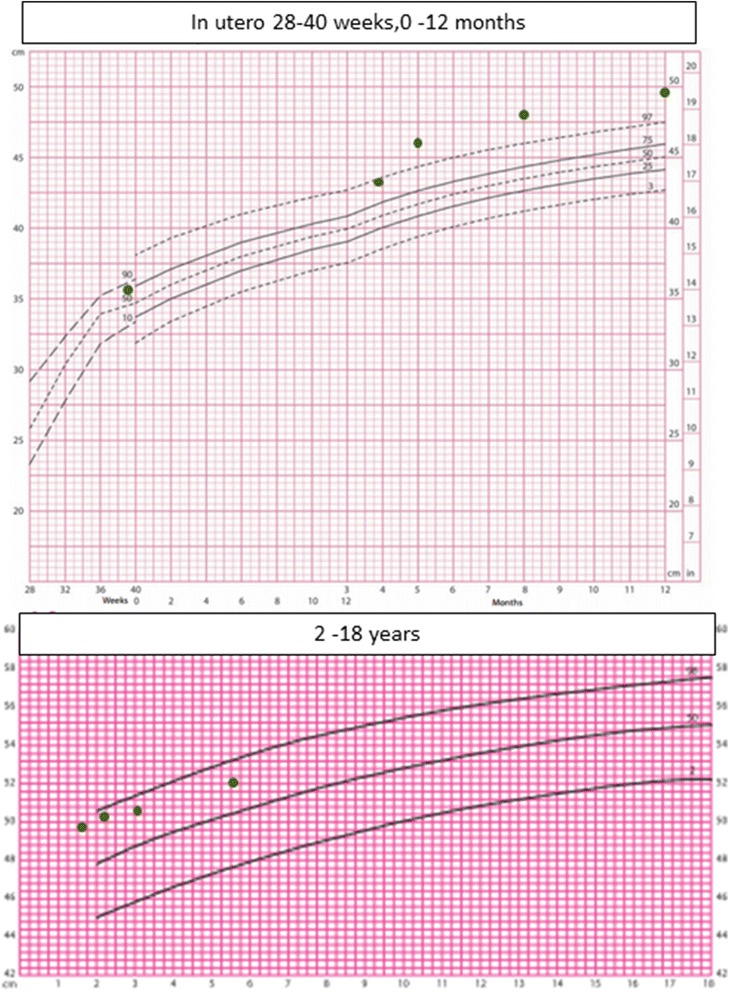
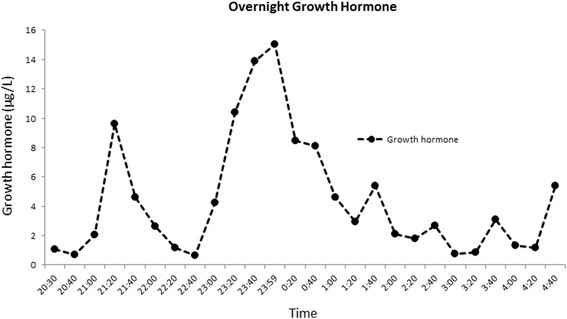
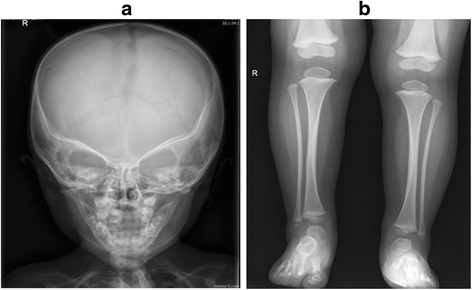
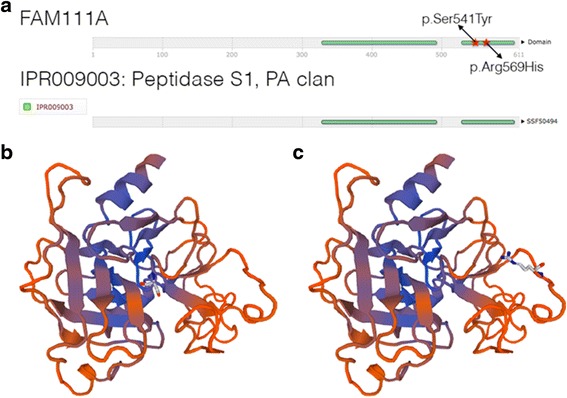
Case presentation: The proband presented in the neonatal period with hypocalcemic seizures secondary to hypoparathyroidism. Her phenotype included small hands and feet, hypoplastic and dystrophic nails, hypoplastic mid-face and macrocrania. Postnatal growth was delayed but neurodevelopment was normal. A skeletal survey at 2 years of age was suggestive of KCS Type 2 and genetic testing revealed a novel de novo heterozygous mutation c.1622C > A (p.Ser541Tyr) in FAM111A. At 3 years and 2 months, her height was 80cms (SDS -3.86). She had normal overnight GH levels. GH therapy was commenced at a dose of 4.9 mg/m2/week for her short stature and low height velocity of 5cms/year. At the end of the first and second years of GH treatment, height velocity was 6.5cms/year and 7.2cms/year, respectively with maximal dose of 7.24 mg/m2/week.
Conclusion: This case highlights the phenotype and the limited response to GH in a child with genetically proven KCS type 2. Long-term registries monitoring growth outcomes following GH therapy in patients with rare genetic conditions may help guide clinical decisions regarding the use and doses of GH in these conditions.
Keywords: FAM111A gene; Genetics; Growth hormone; Hypoparathyroidism; Kenny-Caffey syndrome Type 2; Short stature.
Figures
Similar articles
-
Case report: Late middle-aged features of FAM111A variant, Kenny-Caffey syndrome type 2-suggestive symptoms during a long follow-up.Front Endocrinol (Lausanne). 2023 Jan 4;13:1073173. doi: 10.3389/fendo.2022.1073173. eCollection 2022. Front Endocrinol (Lausanne). 2023. PMID: 36686468 Free PMC article. Review.
-
Compound Heterozygous Variants in FAM111A Cause Autosomal Recessive Kenny-Caffey Syndrome Type 2.J Clin Res Pediatr Endocrinol. 2023 Feb 27;15(1):97-102. doi: 10.4274/jcrpe.galenos.2021.2020.0315. Epub 2021 Aug 12. J Clin Res Pediatr Endocrinol. 2023. PMID: 34382758 Free PMC article.
-
A recurrent de novo FAM111A mutation causes Kenny-Caffey syndrome type 2.J Bone Miner Res. 2014 Apr;29(4):992-8. doi: 10.1002/jbmr.2091. J Bone Miner Res. 2014. PMID: 23996431
-
Further delineation of phenotype and genotype of Kenny-Caffey syndrome type 2 (phenotype and genotype of KCS type 2).Mol Genet Genomic Med. 2024 Apr;12(4):e2433. doi: 10.1002/mgg3.2433. Mol Genet Genomic Med. 2024. PMID: 38591167 Free PMC article. Review.
-
[Kenny-Caffey syndrome and its related syndromes].Nihon Rinsho. 2015 Nov;73(11):1959-64. Nihon Rinsho. 2015. PMID: 26619675 Review. Japanese.
Cited by
-
Gene-nutrient interactions that impact magnesium homeostasis increase risk for neural tube defects in mice exposed to dolutegravir.Front Cell Dev Biol. 2023 Jun 12;11:1175917. doi: 10.3389/fcell.2023.1175917. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37377737 Free PMC article.
-
Functions and evolution of FAM111 serine proteases.Front Mol Biosci. 2022 Dec 15;9:1081166. doi: 10.3389/fmolb.2022.1081166. eCollection 2022. Front Mol Biosci. 2022. PMID: 36589246 Free PMC article. Review.
-
FAM111 protease activity undermines cellular fitness and is amplified by gain-of-function mutations in human disease.EMBO Rep. 2020 Oct 5;21(10):e50662. doi: 10.15252/embr.202050662. Epub 2020 Aug 9. EMBO Rep. 2020. PMID: 32776417 Free PMC article.
-
Case report: Late middle-aged features of FAM111A variant, Kenny-Caffey syndrome type 2-suggestive symptoms during a long follow-up.Front Endocrinol (Lausanne). 2023 Jan 4;13:1073173. doi: 10.3389/fendo.2022.1073173. eCollection 2022. Front Endocrinol (Lausanne). 2023. PMID: 36686468 Free PMC article. Review.
-
FAM111A protects replication forks from protein obstacles via its trypsin-like domain.Nat Commun. 2020 Mar 12;11(1):1318. doi: 10.1038/s41467-020-15170-7. Nat Commun. 2020. PMID: 32165630 Free PMC article.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
