

Novel Compound Heterozygous Mutations Expand the Recognized Phenotypes of FARS2-Linked Disease
- PMID: 27095821
- PMCID: PMC4981184
- DOI: 10.1177/0883073816643402
Novel Compound Heterozygous Mutations Expand the Recognized Phenotypes of FARS2-Linked Disease
Abstract
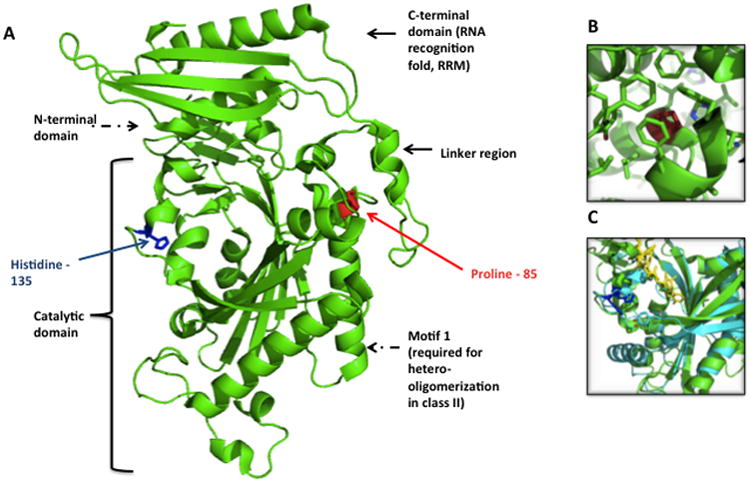
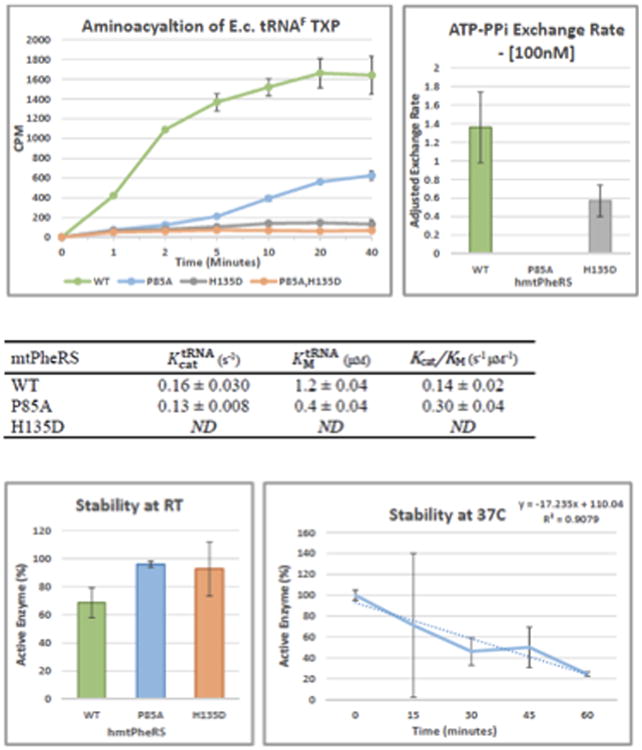
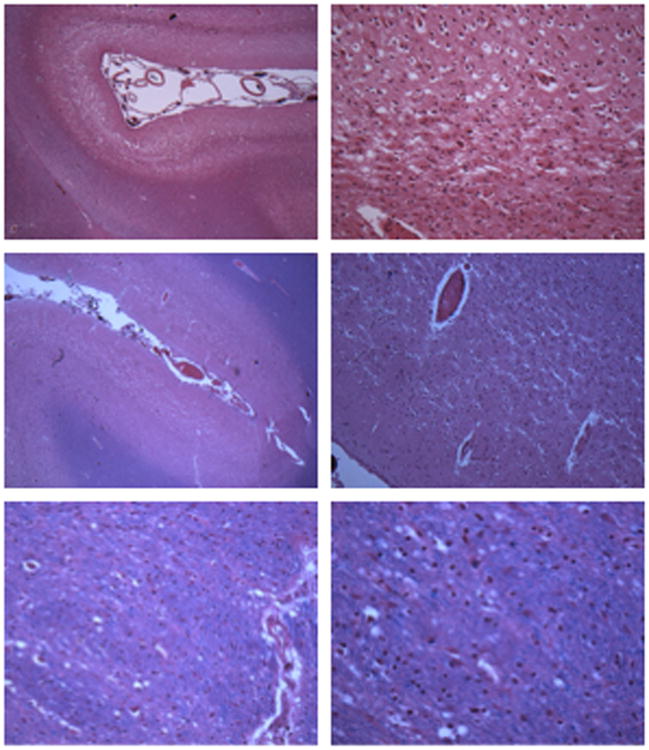
Mutations in mitochondrial aminoacyl-tRNA synthetases are an increasingly recognized cause of human diseases, often arising in individuals with compound heterozygous mutations and presenting with system-specific phenotypes, frequently neurologic. FARS2 encodes mitochondrial phenylalanyl transfer ribonucleic acid (RNA) synthetase (mtPheRS), perturbations of which have been reported in 6 cases of an infantile, lethal disease with refractory epilepsy and progressive myoclonus. Here the authors report the case of juvenile onset refractory epilepsy and progressive myoclonus with compound heterozygous FARS2 mutations. The authors describe the clinical course over 6 years of care at their institution and diagnostic studies including electroencephalogram (EEG), brain magnetic resonance imaging (MRI), serum and cerebrospinal fluid analyses, skeletal muscle biopsy histology, and autopsy gross and histologic findings, which include features shared with Alpers-Huttenlocher syndrome, Leigh syndrome, and a previously published case of FARS2 mutation associated infantile onset disease. The authors also present structure-guided analysis of the relevant mutations based on published mitochondrial phenylalanyl transfer RNA synthetase and related protein crystal structures as well as biochemical analysis of the corresponding recombinant mutant proteins.
Keywords: FARS2; mitochondrial tRNA synthetase; progressive myoclonus epilepsy.
© The Author(s) 2016.
Figures


Similar articles
-
A patient with juvenile-onset refractory status epilepticus caused by two novel compound heterozygous mutations in FARS2 gene.Int J Neurosci. 2019 Nov;129(11):1094-1097. doi: 10.1080/00207454.2019.1634071. Epub 2019 Aug 20. Int J Neurosci. 2019. PMID: 31329004
-
Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy.Hum Mol Genet. 2012 Oct 15;21(20):4521-9. doi: 10.1093/hmg/dds294. Epub 2012 Jul 23. Hum Mol Genet. 2012. PMID: 22833457
-
Clinical findings in a patient with FARS2 mutations and early-infantile-encephalopathy with epilepsy.Am J Med Genet A. 2016 Nov;170(11):3004-3007. doi: 10.1002/ajmg.a.37836. Epub 2016 Aug 23. Am J Med Genet A. 2016. PMID: 27549011
-
FARS2 mutation and epilepsy: Possible link with early-onset epileptic encephalopathy.Epilepsy Res. 2017 Jan;129:118-124. doi: 10.1016/j.eplepsyres.2016.11.022. Epub 2016 Dec 2. Epilepsy Res. 2017. PMID: 28043061 Review.
-
Adult-onset combined oxidative phosphorylation deficiency type 14 manifests as epileptic status: a new phenotype and literature review.BMC Neurol. 2024 Jan 2;24(1):15. doi: 10.1186/s12883-023-03480-4. BMC Neurol. 2024. PMID: 38166857 Free PMC article. Review.
Cited by
-
Clinical Attributes and Electroencephalogram Analysis of Patients With Varying Alpers' Syndrome Genotypes.Front Pharmacol. 2021 Oct 6;12:669516. doi: 10.3389/fphar.2021.669516. eCollection 2021. Front Pharmacol. 2021. PMID: 34690748 Free PMC article.
-
Predictive modeling provides insight into the clinical heterogeneity associated with TARS1 loss-of-function mutations.bioRxiv [Preprint]. 2024 Mar 27:2024.03.25.586600. doi: 10.1101/2024.03.25.586600. bioRxiv. 2024. Update in: HGG Adv. 2024 Jul 18;5(3):100324. doi: 10.1016/j.xhgg.2024.100324. PMID: 38585737 Free PMC article. Updated. Preprint.
-
Two Chinese siblings of combined oxidative phosphorylation deficiency 14 caused by compound heterozygous variants in FARS2.Eur J Med Res. 2022 Sep 26;27(1):184. doi: 10.1186/s40001-022-00808-7. Eur J Med Res. 2022. PMID: 36155627 Free PMC article.
-
When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases.J Biol Chem. 2019 Apr 5;294(14):5309-5320. doi: 10.1074/jbc.REV118.002953. Epub 2019 Jan 15. J Biol Chem. 2019. PMID: 30647134 Free PMC article. Review.
-
Progressive Myoclonus Epilepsy: A Scoping Review of Diagnostic, Phenotypic and Therapeutic Advances.Genes (Basel). 2024 Jan 27;15(2):171. doi: 10.3390/genes15020171. Genes (Basel). 2024. PMID: 38397161 Free PMC article.
References
-
- Konovalova S, Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metabol. 2013;108:206–211. - PubMed
-
- Hallmann K, Zsurka G, Moskau-Hartmann S, Kirschner J, Korinthenberg R, Ruppert AK, Ozdemir O, Weber Y, Becker F, Lerche H, Elger CE, Thiele H, Nürnberg P, Sander T, Kunz WS. A homozygous splice-site mutation in CARS2 is associated with progressive myoclonic epilepsy. Neurology. 2014;83:2183–7. - PubMed
-
- Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, Muravina TI, Serkov SV, Uziel G, Bugiani M, Schiffmann R, Krägeloh-Mann I, Smeitink JA, Florentz C, Van Coster R, Pronk JC, van der Knaap MS. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534–9. - PubMed
-
- Steenweg ME, Ghezzi D, Haack T, Abbink TE, Martinelli D, van Berkel CG, Bley A, Diogo L, Grillo E, Te Water Naudé J, Strom TM, Bertini E, Prokisch H, van der Knaap MS, Zeviani M. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–94. - PubMed
-
- Bayat V, Thiffault I, Jaiswal M, Tétreault M, Donti T, Sasarman F, Bernard G, Demers-Lamarche J, Dicaire MJ, Mathieu J, Vanasse M, Bouchard JP, Rioux MF, Lourenco CM, Li Z, Haueter C, Shoubridge EA, Graham BH, Brais B, Bellen HJ. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10(3):e1001288. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
