

Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement
- PMID: 26000322
- PMCID: PMC4435704
- DOI: 10.1002/acn3.189
Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement
Abstract
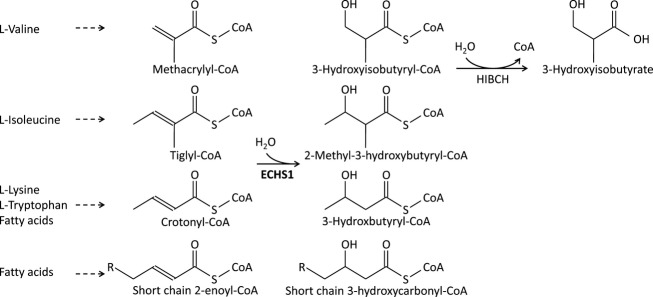
Objective: Short-chain enoyl-CoA hydratase (ECHS1) is a multifunctional mitochondrial matrix enzyme that is involved in the oxidation of fatty acids and essential amino acids such as valine. Here, we describe the broad phenotypic spectrum and pathobiochemistry of individuals with autosomal-recessive ECHS1 deficiency.
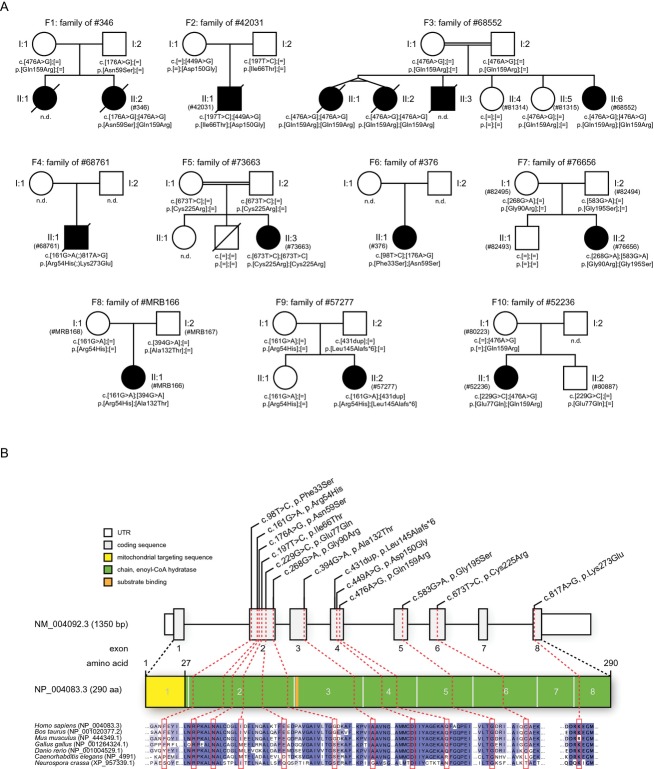
Methods: Using exome sequencing, we identified ten unrelated individuals carrying compound heterozygous or homozygous mutations in ECHS1. Functional investigations in patient-derived fibroblast cell lines included immunoblotting, enzyme activity measurement, and a palmitate loading assay.
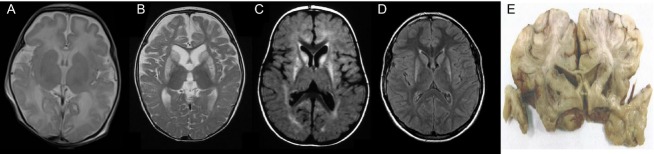
Results: Patients showed a heterogeneous phenotype with disease onset in the first year of life and course ranging from neonatal death to survival into adulthood. The most prominent clinical features were encephalopathy (10/10), deafness (9/9), epilepsy (6/9), optic atrophy (6/10), and cardiomyopathy (4/10). Serum lactate was elevated and brain magnetic resonance imaging showed white matter changes or a Leigh-like pattern resembling disorders of mitochondrial energy metabolism. Analysis of patients' fibroblast cell lines (6/10) provided further evidence for the pathogenicity of the respective mutations by showing reduced ECHS1 protein levels and reduced 2-enoyl-CoA hydratase activity. While serum acylcarnitine profiles were largely normal, in vitro palmitate loading of patient fibroblasts revealed increased butyrylcarnitine, unmasking the functional defect in mitochondrial β-oxidation of short-chain fatty acids. Urinary excretion of 2-methyl-2,3-dihydroxybutyrate - a potential derivative of acryloyl-CoA in the valine catabolic pathway - was significantly increased, indicating impaired valine oxidation.
Interpretation: In conclusion, we define the phenotypic spectrum of a new syndrome caused by ECHS1 deficiency. We speculate that both the β-oxidation defect and the block in l-valine metabolism, with accumulation of toxic methacrylyl-CoA and acryloyl-CoA, contribute to the disorder that may be amenable to metabolic treatment approaches.
Figures

Similar articles
-
ECHS1 deficiency and its biochemical and clinical phenotype.Am J Med Genet A. 2022 Oct;188(10):2908-2919. doi: 10.1002/ajmg.a.62895. Epub 2022 Jul 20. Am J Med Genet A. 2022. PMID: 35856138
-
Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency.Am J Med Genet A. 2018 May;176(5):1115-1127. doi: 10.1002/ajmg.a.38658. Epub 2018 Mar 25. Am J Med Genet A. 2018. PMID: 29575569 Free PMC article.
-
ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism.Brain. 2014 Nov;137(Pt 11):2903-8. doi: 10.1093/brain/awu216. Epub 2014 Aug 14. Brain. 2014. PMID: 25125611
-
Pathogenic Biallelic Mutations in ECHS1 in a Case with Short-Chain Enoyl-CoA Hydratase (SCEH) Deficiency-Case Report and Literature Review.Int J Environ Res Public Health. 2022 Feb 13;19(4):2088. doi: 10.3390/ijerph19042088. Int J Environ Res Public Health. 2022. PMID: 35206276 Free PMC article. Review.
-
Clinical, biochemical and metabolic characterization of patients with short-chain enoyl-CoA hydratase(ECHS1) deficiency: two case reports and the review of the literature.BMC Pediatr. 2020 Feb 3;20(1):50. doi: 10.1186/s12887-020-1947-z. BMC Pediatr. 2020. PMID: 32013919 Free PMC article. Review.
Cited by
-
Cascade testing in mitochondrial diseases: a cross-sectional retrospective study.BMC Neurol. 2024 Sep 13;24(1):343. doi: 10.1186/s12883-024-03850-6. BMC Neurol. 2024. PMID: 39272026 Free PMC article.
-
Mechanistic insight into 3-methylmercaptopropionate metabolism and kinetical regulation of demethylation pathway in marine dimethylsulfoniopropionate-catabolizing bacteria.Mol Microbiol. 2019 Apr;111(4):1057-1073. doi: 10.1111/mmi.14211. Epub 2019 Mar 4. Mol Microbiol. 2019. PMID: 30677184 Free PMC article.
-
Valine metabolites analysis in ECHS1 deficiency.Mol Genet Metab Rep. 2021 Oct 9;29:100809. doi: 10.1016/j.ymgmr.2021.100809. eCollection 2021 Dec. Mol Genet Metab Rep. 2021. PMID: 34667719 Free PMC article.
-
ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: case presentation and initial benefit of intervention.J Neurol. 2017 Jan;264(1):185-187. doi: 10.1007/s00415-016-8381-z. Epub 2016 Dec 30. J Neurol. 2017. PMID: 28039521
-
Exercise improves choroid plexus epithelial cells metabolism to prevent glial cell-associated neurodegeneration.Front Pharmacol. 2022 Sep 16;13:1010785. doi: 10.3389/fphar.2022.1010785. eCollection 2022. Front Pharmacol. 2022. PMID: 36188600 Free PMC article.
References
-
- Kanazawa M, Ohtake A, Abe H, et al. Molecular cloning and sequence analysis of the cDNA for human mitochondrial short-chain enoyl-CoA hydratase. Enzyme Protein. 1993;47:9–13. - PubMed
-
- Pougovkina O, te Brinke H, Ofman R, et al. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum Mol Genet. 2014;23:3513–3522. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
