

X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes
- PMID: 25644381
- PMCID: PMC5414091
- DOI: 10.1038/mp.2014.193
X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes
Abstract
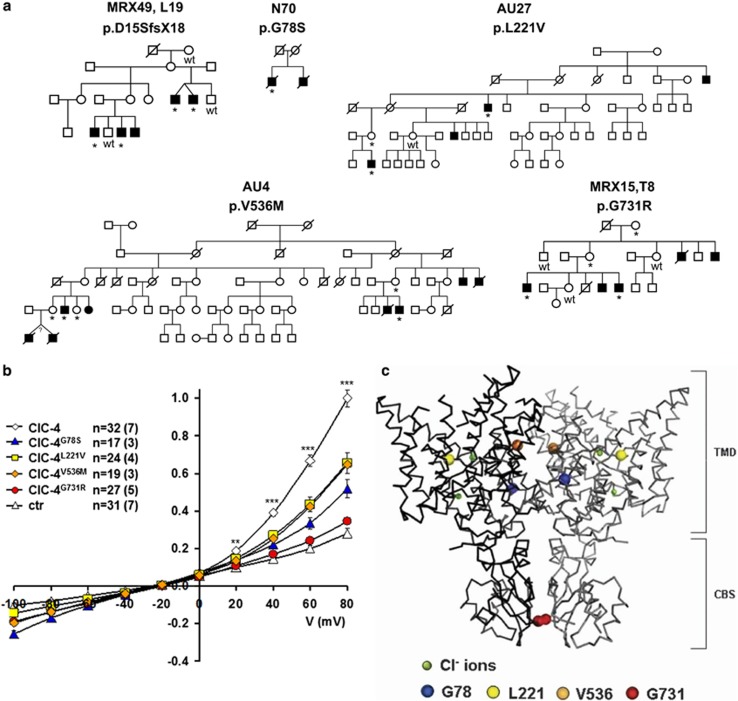
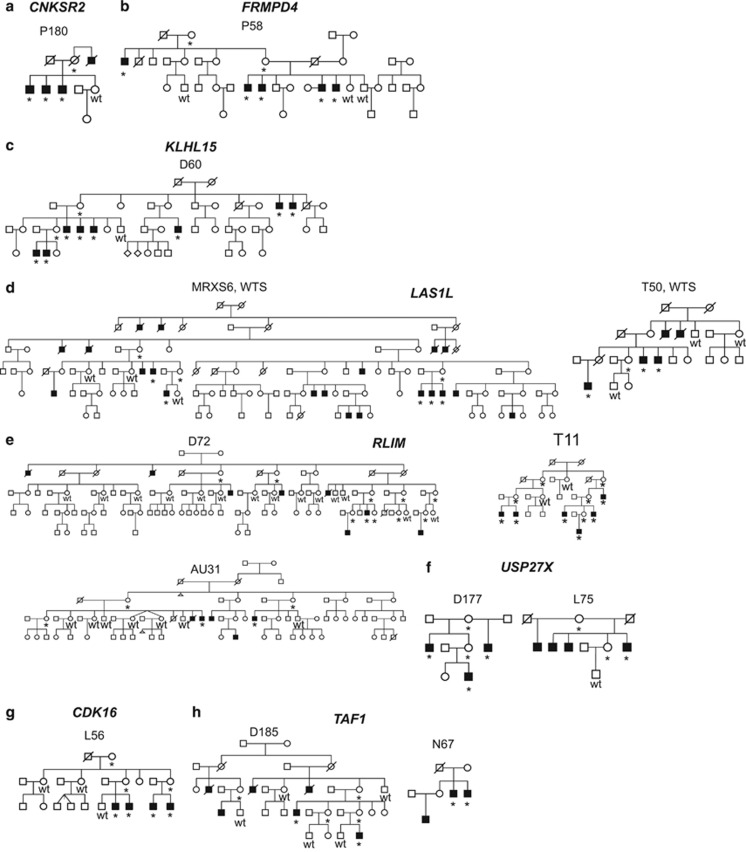
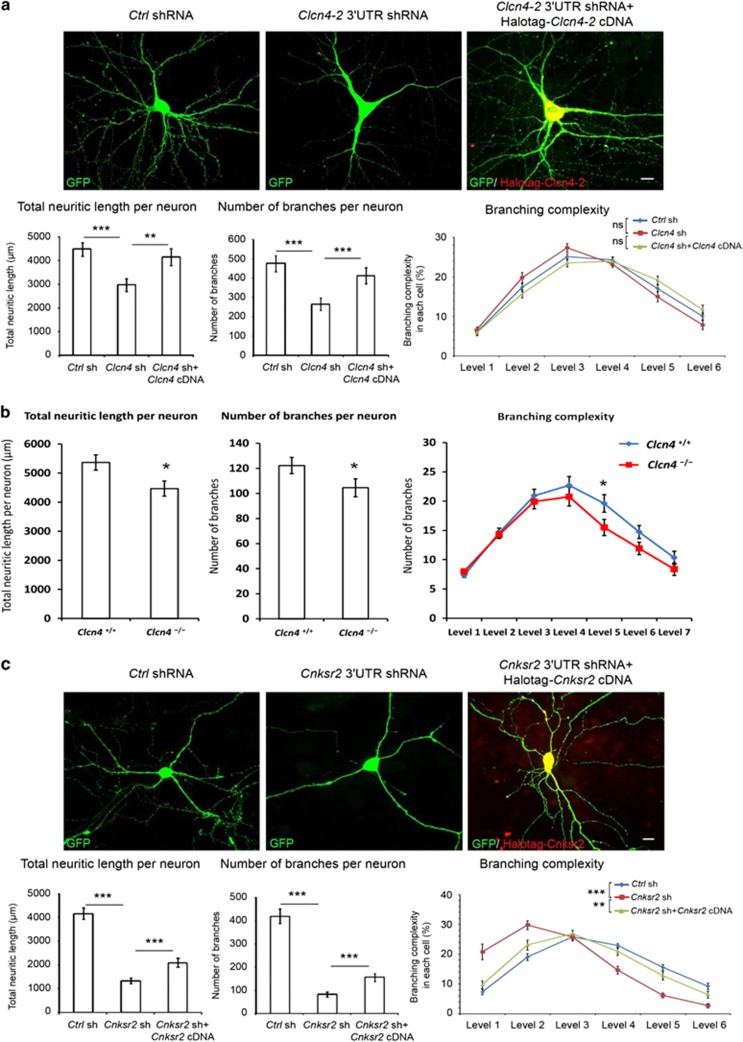
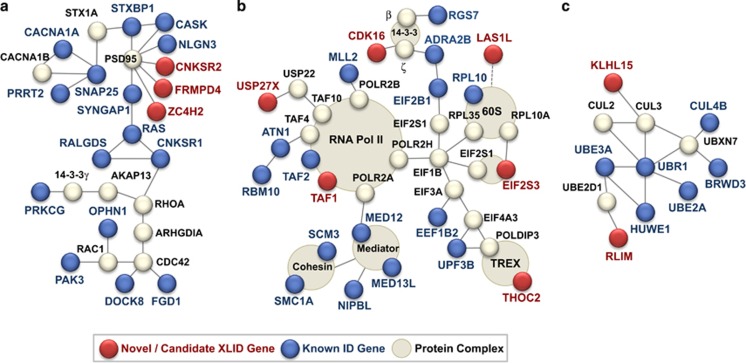
X-linked intellectual disability (XLID) is a clinically and genetically heterogeneous disorder. During the past two decades in excess of 100 X-chromosome ID genes have been identified. Yet, a large number of families mapping to the X-chromosome remained unresolved suggesting that more XLID genes or loci are yet to be identified. Here, we have investigated 405 unresolved families with XLID. We employed massively parallel sequencing of all X-chromosome exons in the index males. The majority of these males were previously tested negative for copy number variations and for mutations in a subset of known XLID genes by Sanger sequencing. In total, 745 X-chromosomal genes were screened. After stringent filtering, a total of 1297 non-recurrent exonic variants remained for prioritization. Co-segregation analysis of potential clinically relevant changes revealed that 80 families (20%) carried pathogenic variants in established XLID genes. In 19 families, we detected likely causative protein truncating and missense variants in 7 novel and validated XLID genes (CLCN4, CNKSR2, FRMPD4, KLHL15, LAS1L, RLIM and USP27X) and potentially deleterious variants in 2 novel candidate XLID genes (CDK16 and TAF1). We show that the CLCN4 and CNKSR2 variants impair protein functions as indicated by electrophysiological studies and altered differentiation of cultured primary neurons from Clcn4(-/-) mice or after mRNA knock-down. The newly identified and candidate XLID proteins belong to pathways and networks with established roles in cognitive function and intellectual disability in particular. We suggest that systematic sequencing of all X-chromosomal genes in a cohort of patients with genetic evidence for X-chromosome locus involvement may resolve up to 58% of Fragile X-negative cases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
[Clinical features and genetic analysis of 17 Chinese pedigrees affected with X-linked intellectual disability].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2024 May 10;41(5):533-539. doi: 10.3760/cma.j.cn511374-20230808-00048. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2024. PMID: 38684296 Chinese.
-
Pathogenic variants in E3 ubiquitin ligase RLIM/RNF12 lead to a syndromic X-linked intellectual disability and behavior disorder.Mol Psychiatry. 2019 Nov;24(11):1748-1768. doi: 10.1038/s41380-018-0065-x. Epub 2018 May 4. Mol Psychiatry. 2019. PMID: 29728705
-
HUWE1 mutations in Juberg-Marsidi and Brooks syndromes: the results of an X-chromosome exome sequencing study.BMJ Open. 2016 Apr 29;6(4):e009537. doi: 10.1136/bmjopen-2015-009537. BMJ Open. 2016. PMID: 27130160 Free PMC article.
-
Non-syndromic X linked intellectual disability: Current knowledge in light of the recent advances in molecular and functional studies.Clin Genet. 2020 May;97(5):677-687. doi: 10.1111/cge.13698. Epub 2020 Jan 9. Clin Genet. 2020. PMID: 31898314 Review.
-
Whole exome sequencing revealed variants in four genes underlying X-linked intellectual disability in four Iranian families: novel deleterious variants and clinical features with the review of literature.BMC Med Genomics. 2023 Oct 11;16(1):239. doi: 10.1186/s12920-023-01680-y. BMC Med Genomics. 2023. PMID: 37821930 Free PMC article. Review.
Cited by
-
A non-coding variant in the 5' UTR of DLG3 attenuates protein translation to cause non-syndromic intellectual disability.Eur J Hum Genet. 2016 Nov;24(11):1612-1616. doi: 10.1038/ejhg.2016.46. Epub 2016 May 25. Eur J Hum Genet. 2016. PMID: 27222290 Free PMC article.
-
Expanding the genetics and phenotypic spectrum of Lysine-specific demethylase 5C (KDM5C): a report of 13 novel variants.Eur J Hum Genet. 2023 Feb;31(2):202-215. doi: 10.1038/s41431-022-01233-4. Epub 2022 Nov 25. Eur J Hum Genet. 2023. PMID: 36434256 Free PMC article.
-
Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies.J Neurochem. 2019 Feb;148(3):325-347. doi: 10.1111/jnc.14576. Epub 2018 Nov 12. J Neurochem. 2019. PMID: 30144322 Free PMC article. Review.
-
SAGA-Dependent Histone H2Bub1 Deubiquitination Is Essential for Cellular Ubiquitin Balance during Embryonic Development.Int J Mol Sci. 2022 Jul 5;23(13):7459. doi: 10.3390/ijms23137459. Int J Mol Sci. 2022. PMID: 35806465 Free PMC article. Review.
-
Intellectual disability genomics: current state, pitfalls and future challenges.BMC Genomics. 2021 Dec 20;22(1):909. doi: 10.1186/s12864-021-08227-4. BMC Genomics. 2021. PMID: 34930158 Free PMC article. Review.
References
-
- Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet 2005; 6: 46–57. - PubMed
-
- de Brouwer AP, Yntema HG, Kleefstra T, Lugtenberg D, Oudakker AR, de Vries BB et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum Mutat 2007; 28: 207–208. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
