

CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria
- PMID: 25597511
- PMCID: PMC4320254
- DOI: 10.1016/j.ajhg.2014.12.020
CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria
Abstract
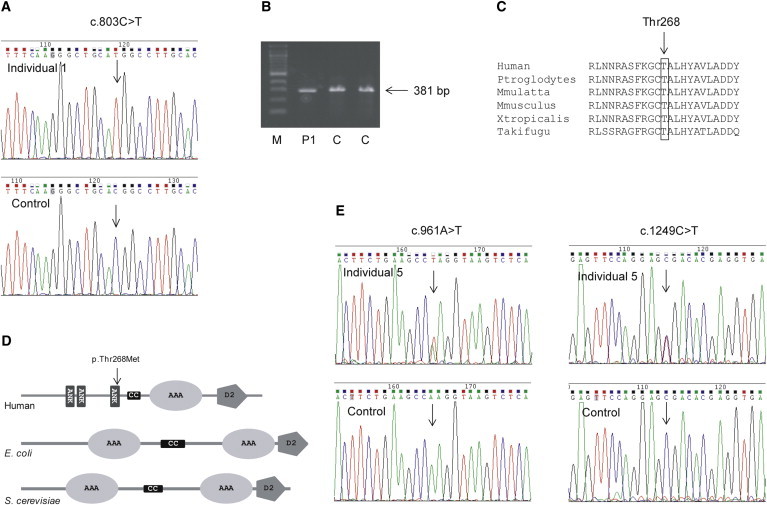
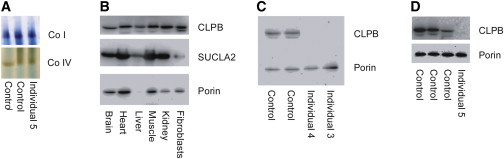
3-methylglutaconic aciduria (3-MGA-uria) is a nonspecific finding associated with mitochondrial dysfunction, including defects of oxidative phosphorylation. 3-MGA-uria is classified into five groups, of which one, type IV, is genetically heterogeneous. Here we report five children with a form of type IV 3-MGA-uria characterized by cataracts, severe psychomotor regression during febrile episodes, epilepsy, neutropenia with frequent infections, and death in early childhood. Four of the individuals were of Greenlandic descent, and one was North American, of Northern European and Asian descent. Through a combination of homozygosity mapping in the Greenlandic individuals and exome sequencing in the North American, we identified biallelic variants in the caseinolytic peptidase B homolog (CLPB). The causative variants included one missense variant, c.803C>T (p.Thr268Met), and two nonsense variants, c.961A>T (p.Lys321*) and c.1249C>T (p.Arg417*). The level of CLPB protein was markedly decreased in fibroblasts and liver of affected individuals. CLPB is proposed to function as a mitochondrial chaperone involved in disaggregation of misfolded proteins, resulting from stress such as heat denaturation.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder.Am J Hum Genet. 2015 Feb 5;96(2):245-57. doi: 10.1016/j.ajhg.2014.12.013. Epub 2015 Jan 15. Am J Hum Genet. 2015. PMID: 25597510 Free PMC article.
-
Bi-allelic CLPB mutations cause cataract, renal cysts, nephrocalcinosis and 3-methylglutaconic aciduria, a novel disorder of mitochondrial protein disaggregation.J Inherit Metab Dis. 2015 Mar;38(2):211-9. doi: 10.1007/s10545-015-9813-0. Epub 2015 Jan 18. J Inherit Metab Dis. 2015. PMID: 25595726
-
HTRA2 Defect: A Recognizable Inborn Error of Metabolism with 3-Methylglutaconic Aciduria as Discriminating Feature Characterized by Neonatal Movement Disorder and Epilepsy-Report of 11 Patients.Neuropediatrics. 2018 Dec;49(6):373-378. doi: 10.1055/s-0038-1667345. Epub 2018 Aug 16. Neuropediatrics. 2018. PMID: 30114719
-
Clinical manifestations and enzymatic activities of mitochondrial respiratory chain complexes in Pearson marrow-pancreas syndrome with 3-methylglutaconic aciduria: a case report and literature review.Eur J Pediatr. 2015 Dec;174(12):1593-602. doi: 10.1007/s00431-015-2576-7. Epub 2015 Jun 16. Eur J Pediatr. 2015. PMID: 26074369 Review.
-
Costeff optic atrophy syndrome: new clinical case and novel molecular findings.J Inherit Metab Dis. 2008 Dec;31 Suppl 2:S419-23. doi: 10.1007/s10545-008-0981-z. Epub 2008 Nov 7. J Inherit Metab Dis. 2008. PMID: 18985435 Review.
Cited by
-
A scoring system predicting the clinical course of CLPB defect based on the foetal and neonatal presentation of 31 patients.J Inherit Metab Dis. 2017 Nov;40(6):853-860. doi: 10.1007/s10545-017-0057-z. Epub 2017 Jul 7. J Inherit Metab Dis. 2017. PMID: 28687938
-
[Leigh syndrome caused by the mitochondrial m.8993T>G mutation with hypocitrullinemia: a report of four cases and literature review].Zhongguo Dang Dai Er Ke Za Zhi. 2024 Sept 15;26(9):940-945. doi: 10.7499/j.issn.1008-8830.2404036. Zhongguo Dang Dai Er Ke Za Zhi. 2024. PMID: 39267509 Free PMC article. Review. Chinese.
-
Heterozygous variants of CLPB are a cause of severe congenital neutropenia.Blood. 2022 Feb 3;139(5):779-791. doi: 10.1182/blood.2021010762. Blood. 2022. PMID: 34115842 Free PMC article.
-
Mitochondrial Protein Homeostasis and Cardiomyopathy.Int J Mol Sci. 2022 Mar 20;23(6):3353. doi: 10.3390/ijms23063353. Int J Mol Sci. 2022. PMID: 35328774 Free PMC article. Review.
-
Comprehensive structural characterization of the human AAA+ disaggregase CLPB in the apo- and substrate-bound states reveals a unique mode of action driven by oligomerization.PLoS Biol. 2023 Feb 6;21(2):e3001987. doi: 10.1371/journal.pbio.3001987. eCollection 2023 Feb. PLoS Biol. 2023. PMID: 36745679 Free PMC article.
References
-
- Gunay-Aygun M. 3-Methylglutaconic aciduria: a common biochemical marker in various syndromes with diverse clinical features. Mol. Genet. Metab. 2005;84:1–3. - PubMed
-
- Marinier E., Lincoln B.C., Garneau M., David F., Brunengraber H. Contribution of the shunt pathway of mevalonate metabolism to the regulation of cholesterol synthesis in rat liver. J. Biol. Chem. 1987;262:16936–16940. - PubMed
-
- Weinstock S.B., Kopito R.R., Endemann G., Tomera J.F., Marinier E., Murray D.M., Brunengraber H. The shunt pathway of mevalonate metabolism in the isolated perfused rat liver. J. Biol. Chem. 1984;259:8939–8944. - PubMed
-
- Wortmann S.B., Duran M., Anikster Y., Barth P.G., Sperl W., Zschocke J., Morava E., Wevers R.A. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J. Inherit. Metab. Dis. 2013;36:923–928. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
