

Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies
- PMID: 25058219
- PMCID: PMC6558267
- DOI: 10.1001/jama.2014.7184
Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies
Abstract
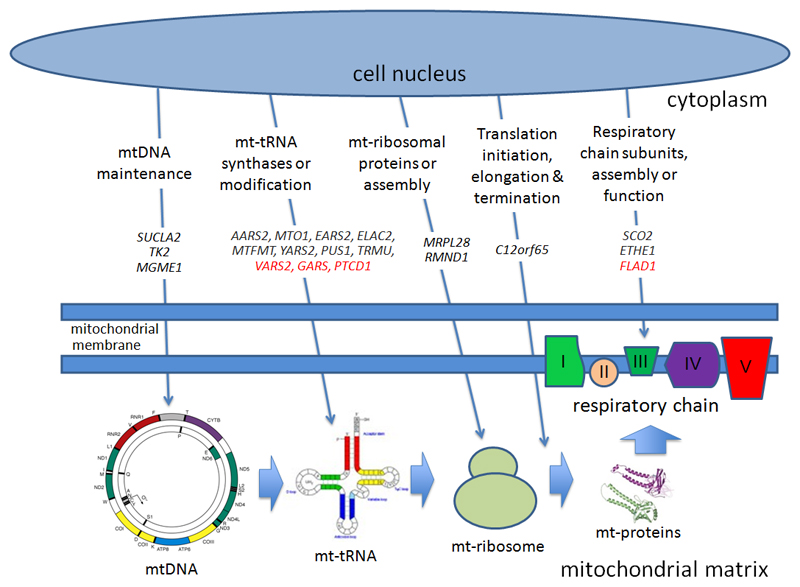
Importance: Mitochondrial disorders have emerged as a common cause of inherited disease, but their diagnosis remains challenging. Multiple respiratory chain complex defects are particularly difficult to diagnose at the molecular level because of the massive number of nuclear genes potentially involved in intramitochondrial protein synthesis, with many not yet linked to human disease.
Objective: To determine the molecular basis of multiple respiratory chain complex deficiencies.
Design, setting, and participants: We studied 53 patients referred to 2 national centers in the United Kingdom and Germany between 2005 and 2012. All had biochemical evidence of multiple respiratory chain complex defects but no primary pathogenic mitochondrial DNA mutation. Whole-exome sequencing was performed using 62-Mb exome enrichment, followed by variant prioritization using bioinformatic prediction tools, variant validation by Sanger sequencing, and segregation of the variant with the disease phenotype in the family.
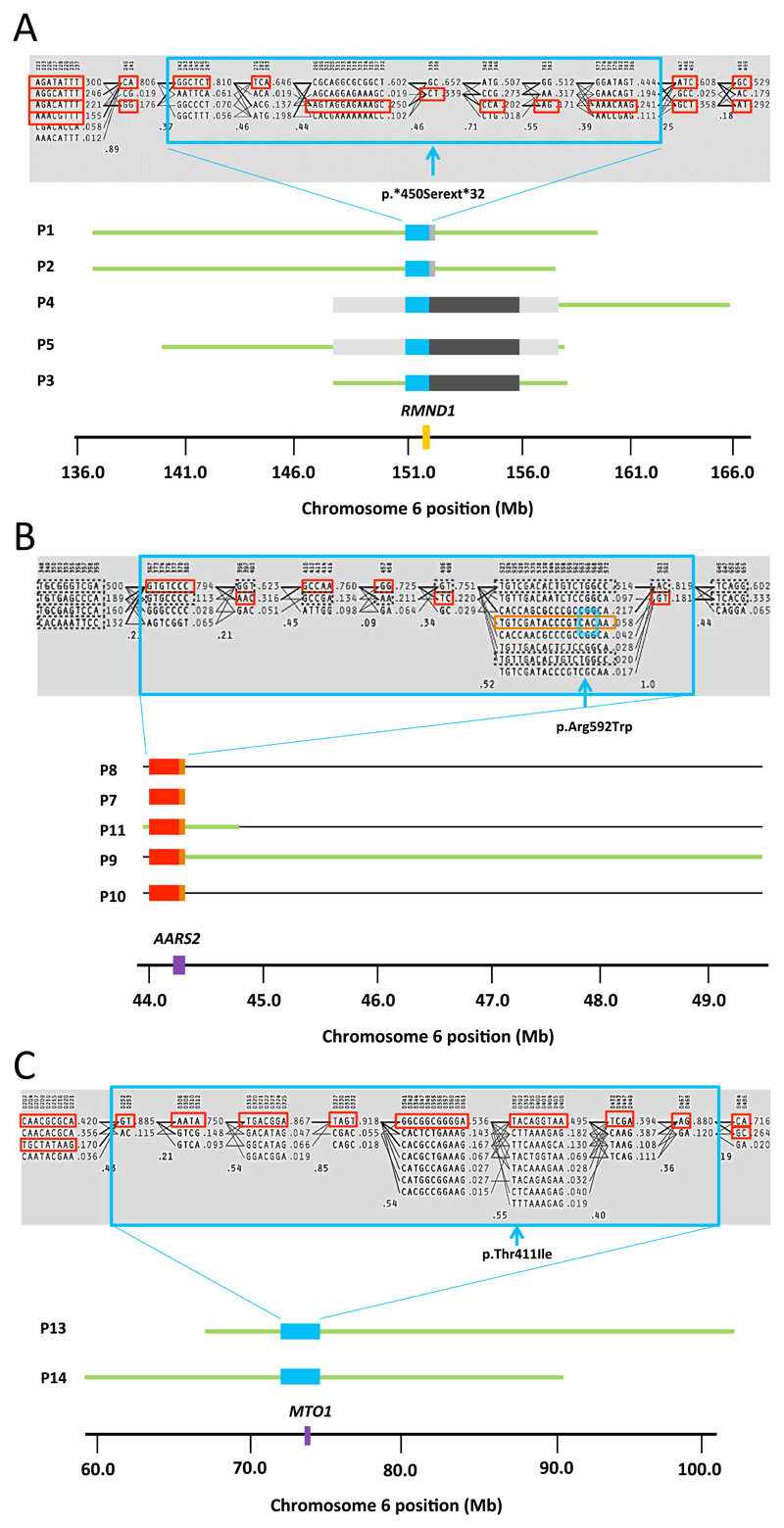
Results: Presumptive causal variants were identified in 28 patients (53%; 95% CI, 39%-67%) and possible causal variants were identified in 4 (8%; 95% CI, 2%-18%). Together these accounted for 32 patients (60% 95% CI, 46%-74%) and involved 18 different genes. These included recurrent mutations in RMND1, AARS2, and MTO1, each on a haplotype background consistent with a shared founder allele, and potential novel mutations in 4 possible mitochondrial disease genes (VARS2, GARS, FLAD1, and PTCD1). Distinguishing clinical features included deafness and renal involvement associated with RMND1 and cardiomyopathy with AARS2 and MTO1. However, atypical clinical features were present in some patients, including normal liver function and Leigh syndrome (subacute necrotizing encephalomyelopathy) seen in association with TRMU mutations and no cardiomyopathy with founder SCO2 mutations. It was not possible to confidently identify the underlying genetic basis in 21 patients (40%; 95% CI, 26%-54%).
Conclusions and relevance: Exome sequencing enhances the ability to identify potential nuclear gene mutations in patients with biochemically defined defects affecting multiple mitochondrial respiratory chain complexes. Additional study is required in independent patient populations to determine the utility of this approach in comparison with traditional diagnostic methods.
Conflict of interest statement
The authors report no conflicts of interest
Figures
Similar articles
-
New perspective in diagnostics of mitochondrial disorders: two years' experience with whole-exome sequencing at a national paediatric centre.J Transl Med. 2016 Jun 12;14(1):174. doi: 10.1186/s12967-016-0930-9. J Transl Med. 2016. PMID: 27290639 Free PMC article.
-
Targeted exome sequencing of suspected mitochondrial disorders.Neurology. 2013 May 7;80(19):1762-70. doi: 10.1212/WNL.0b013e3182918c40. Epub 2013 Apr 17. Neurology. 2013. PMID: 23596069 Free PMC article.
-
A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies.PLoS Genet. 2016 Jan 7;12(1):e1005679. doi: 10.1371/journal.pgen.1005679. eCollection 2016 Jan. PLoS Genet. 2016. PMID: 26741492 Free PMC article.
-
Diagnosis and molecular basis of mitochondrial respiratory chain disorders: exome sequencing for disease gene identification.Biochim Biophys Acta. 2014 Apr;1840(4):1355-9. doi: 10.1016/j.bbagen.2014.01.025. Epub 2014 Jan 24. Biochim Biophys Acta. 2014. PMID: 24462578 Review.
-
Molecular genetic testing for mitochondrial disease: from one generation to the next.Neurotherapeutics. 2013 Apr;10(2):251-61. doi: 10.1007/s13311-012-0174-1. Neurotherapeutics. 2013. PMID: 23269497 Free PMC article. Review.
Cited by
-
Recessive Mutations in TRMT10C Cause Defects in Mitochondrial RNA Processing and Multiple Respiratory Chain Deficiencies.Am J Hum Genet. 2016 May 5;98(5):993-1000. doi: 10.1016/j.ajhg.2016.03.010. Epub 2016 Apr 28. Am J Hum Genet. 2016. PMID: 27132592 Free PMC article.
-
Mechanisms and Future Research Perspectives on Mitochondrial Diseases Associated with Isoleucyl-tRNA Synthetase Gene Mutations.Genes (Basel). 2024 Jul 8;15(7):894. doi: 10.3390/genes15070894. Genes (Basel). 2024. PMID: 39062673 Free PMC article. Review.
-
A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation.Cell Metab. 2016 Dec 13;24(6):875-885. doi: 10.1016/j.cmet.2016.08.017. Epub 2016 Sep 22. Cell Metab. 2016. PMID: 27667664 Free PMC article.
-
Mitochondrial Dysfunction as an Underlying Cause of Skeletal Muscle Disorders.Int J Mol Sci. 2022 Oct 26;23(21):12926. doi: 10.3390/ijms232112926. Int J Mol Sci. 2022. PMID: 36361713 Free PMC article. Review.
-
Unveiling novel genetic variants in 370 challenging medically relevant genes using the long read sequencing data of 41 samples from 19 global populations.Mol Genet Genomics. 2024 Jul 7;299(1):65. doi: 10.1007/s00438-024-02158-x. Mol Genet Genomics. 2024. PMID: 38972030
References
-
- Skladal D, Bernier FP, Halliday JL, Thorburn DR. Birth prevalence of mitochondrial respiratory chain defects in children. J Inher Metab Dis. 2000;23:138.
-
- Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med. 2012;44:41–59. - PubMed
-
- McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–40. - PubMed
-
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–83. - PubMed
Publication types
MeSH terms
Grants and funding
- MR/K000608/1/MRC_/Medical Research Council/United Kingdom
- G1000848/MRC_/Medical Research Council/United Kingdom
- 101876/Z/13/Z/WT_/Wellcome Trust/United Kingdom
- 309548/ERC_/European Research Council/International
- MR/K006312/1/MRC_/Medical Research Council/United Kingdom
- G0601943/MRC_/Medical Research Council/United Kingdom
- 096919Z/11/Z/WT_/Wellcome Trust/United Kingdom
- 101876/WT_/Wellcome Trust/United Kingdom
- G0800674/MRC_/Medical Research Council/United Kingdom
- 096919/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- NIHR-HCS-D12-03-04/DH_/Department of Health/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
