

Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH
- PMID: 24836451
- PMCID: PMC4121479
- DOI: 10.1016/j.ajhg.2014.04.015
Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH
Abstract
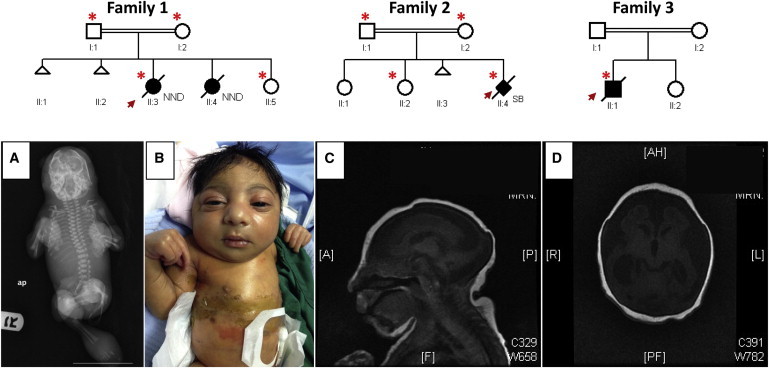
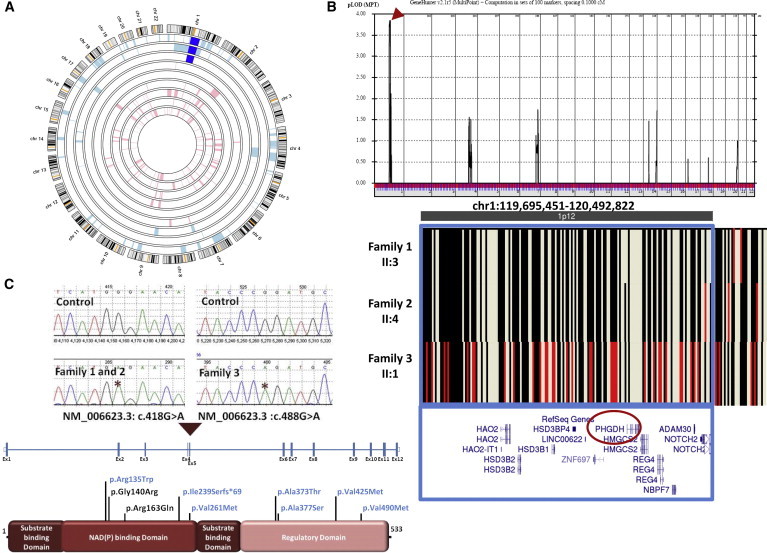
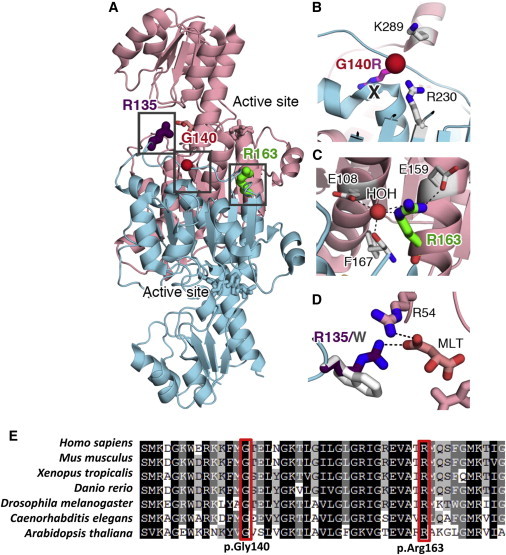
Neu-Laxova syndrome (NLS) is a rare autosomal-recessive disorder characterized by severe fetal growth restriction, microcephaly, a distinct facial appearance, ichthyosis, skeletal anomalies, and perinatal lethality. The pathogenesis of NLS remains unclear despite extensive clinical and pathological phenotyping of the >70 affected individuals reported to date, emphasizing the need to identify the underlying genetic etiology, which remains unknown. In order to identify the cause of NLS, we conducted a positional-mapping study combining autozygosity mapping and whole-exome sequencing in three consanguineous families affected by NLS. Surprisingly, the NLS-associated locus identified in this study was solved at the gene level to reveal mutations in PHGDH, which is known to be mutated in individuals with microcephaly and developmental delay. PHGDH encodes the first enzyme in the phosphorylated pathway of de novo serine synthesis, and complete deficiency of its mouse ortholog recapitulates many of the key features of NLS. This study shows that NLS represents the extreme end of a known inborn error of serine metabolism and highlights the power of genomic sequencing in revealing the unsuspected allelic nature of apparently distinct clinical entities.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Identification of a premature stop codon mutation in the PHGDH gene in severe Neu-Laxova syndrome-evidence for phenotypic variability.Am J Med Genet A. 2015 Jun;167(6):1323-9. doi: 10.1002/ajmg.a.36930. Epub 2015 Apr 25. Am J Med Genet A. 2015. PMID: 25913727
-
Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway.Am J Hum Genet. 2014 Sep 4;95(3):285-93. doi: 10.1016/j.ajhg.2014.07.012. Epub 2014 Aug 21. Am J Hum Genet. 2014. PMID: 25152457 Free PMC article.
-
Novel and recurrent PHGDH and PSAT1 mutations in Chinese patients with Neu-Laxova syndrome.Eur J Dermatol. 2019 Dec 1;29(6):641-646. doi: 10.1684/ejd.2019.3673. Eur J Dermatol. 2019. PMID: 31903955
-
[Neu-Laxova syndrome: Three case reports and a review of the literature].Ann Pathol. 2016 Aug;36(4):235-44. doi: 10.1016/j.annpat.2016.04.004. Epub 2016 Jul 27. Ann Pathol. 2016. PMID: 27475004 Review. French.
-
Mild phenotypes of phosphoglycerate dehydrogenase deficiency by a novel mutation of PHGDH gene: Case report and literature review.Int J Dev Neurosci. 2023 Feb;83(1):44-52. doi: 10.1002/jdn.10236. Epub 2022 Nov 9. Int J Dev Neurosci. 2023. PMID: 36308023 Review.
Cited by
-
Molecular autopsy in maternal-fetal medicine.Genet Med. 2018 Apr;20(4):420-427. doi: 10.1038/gim.2017.111. Epub 2017 Jul 20. Genet Med. 2018. PMID: 28749478
-
Serine biosynthesis as a novel therapeutic target for dilated cardiomyopathy.Eur Heart J. 2022 Sep 21;43(36):3477-3489. doi: 10.1093/eurheartj/ehac305. Eur Heart J. 2022. PMID: 35728000 Free PMC article.
-
On the phenotypic spectrum of serine biosynthesis defects.J Inherit Metab Dis. 2016 May;39(3):373-381. doi: 10.1007/s10545-016-9921-5. Epub 2016 Mar 10. J Inherit Metab Dis. 2016. PMID: 26960553
-
Clinical and genetic heterogeneity of primary ciliopathies (Review).Int J Mol Med. 2021 Sep;48(3):176. doi: 10.3892/ijmm.2021.5009. Epub 2021 Jul 19. Int J Mol Med. 2021. PMID: 34278440 Free PMC article. Review.
-
Discovery of mutations for Mendelian disorders.Hum Genet. 2016 Jun;135(6):615-23. doi: 10.1007/s00439-016-1664-8. Epub 2016 Apr 11. Hum Genet. 2016. PMID: 27068822 Review.
References
-
- Neu R.L., Kajii T., Gardner L.I., Nagyfy S.F. A lethal syndrome of microcephaly with multiple congenital anomalies in three siblings. Pediatrics. 1971;47:610–612. - PubMed
-
- Laxova R., Ohara P.T., Timothy J.A. A further example of a lethal autosomal recessive condition in sibs. J. Ment. Defic. Res. 1972;16:139–143. - PubMed
-
- Lazjuk G.I., Lurie I.W., Ostrowskaja T.I., Cherstvoy E.D., Kirillova I.A., Nedzved M.K., Usoev S.S. Brief clinical observations: the Neu-Laxova syndrome—a distinct entity. Am. J. Med. Genet. 1979;3:261–267. - PubMed
-
- Ostrovskaya T.I., Lazjuk G.I. Cerebral abnormalities in the Neu-Laxova syndrome. Am. J. Med. Genet. 1988;30:747–756. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
