

Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance
- PMID: 23993193
- PMCID: PMC3769921
- DOI: 10.1016/j.ajhg.2013.07.017
Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance
Erratum in
- Am J Hum Genet. 2013 Oct 3;93(4):773
Abstract
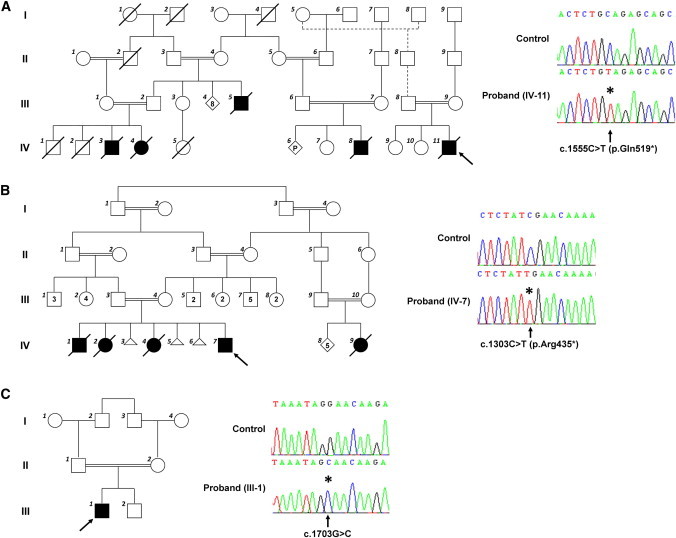
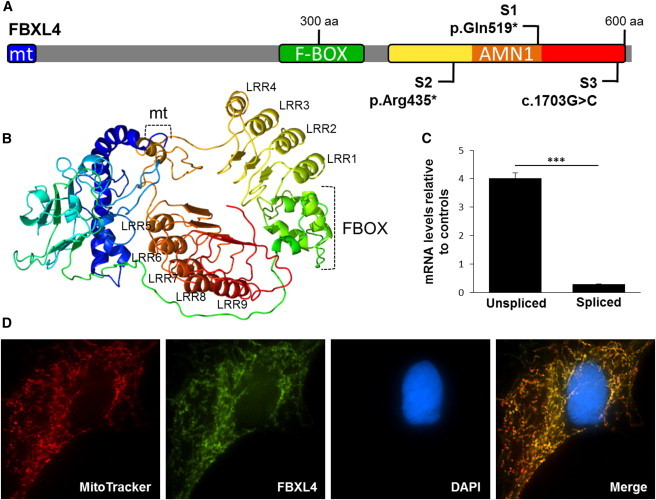
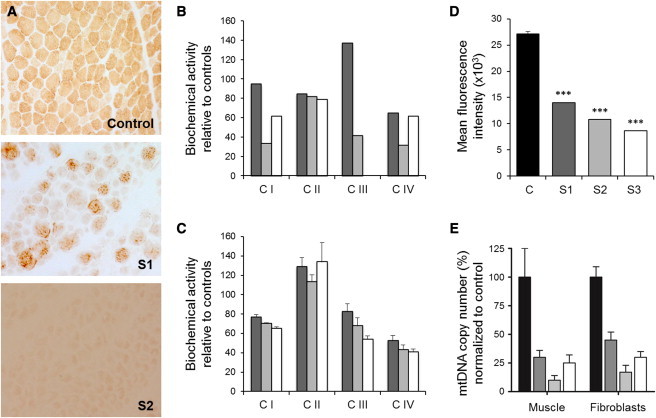
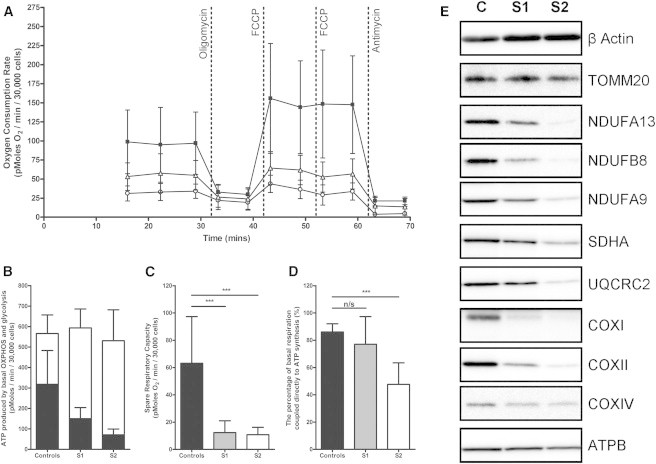
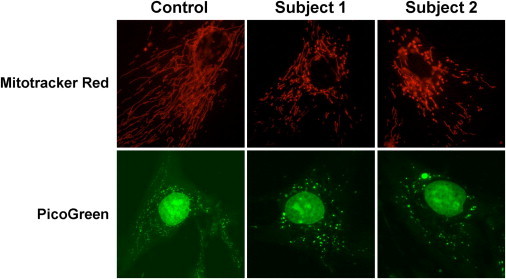
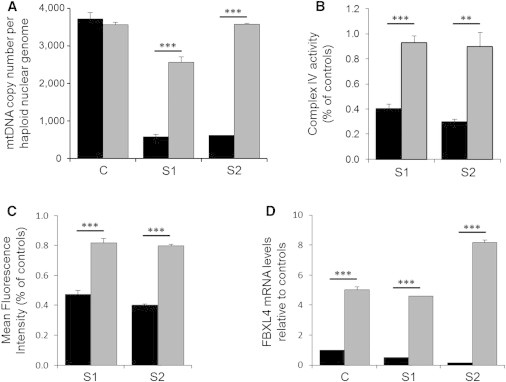
Nuclear genetic disorders causing mitochondrial DNA (mtDNA) depletion are clinically and genetically heterogeneous, and the molecular etiology remains undiagnosed in the majority of cases. Through whole-exome sequencing, we identified recessive nonsense and splicing mutations in FBXL4 segregating in three unrelated consanguineous kindreds in which affected children present with a fatal encephalopathy, lactic acidosis, and severe mtDNA depletion in muscle. We show that FBXL4 is an F-box protein that colocalizes with mitochondria and that loss-of-function and splice mutations in this protein result in a severe respiratory chain deficiency, loss of mitochondrial membrane potential, and a disturbance of the dynamic mitochondrial network and nucleoid distribution in fibroblasts from affected individuals. Expression of the wild-type FBXL4 transcript in cell lines from two subjects fully rescued the levels of mtDNA copy number, leading to a correction of the mitochondrial biochemical deficit. Together our data demonstrate that mutations in FBXL4 are disease causing and establish FBXL4 as a mitochondrial protein with a possible role in maintaining mtDNA integrity and stability.
Copyright © 2013 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy.Am J Hum Genet. 2013 Sep 5;93(3):482-95. doi: 10.1016/j.ajhg.2013.07.016. Epub 2013 Aug 29. Am J Hum Genet. 2013. PMID: 23993194 Free PMC article.
-
A novel mutation in FBXL4 in a Norwegian child with encephalomyopathic mitochondrial DNA depletion syndrome 13.Eur J Med Genet. 2016 Jun;59(6-7):342-6. doi: 10.1016/j.ejmg.2016.05.005. Epub 2016 May 13. Eur J Med Genet. 2016. PMID: 27182039
-
Molecular and clinical spectra of FBXL4 deficiency.Hum Mutat. 2017 Dec;38(12):1649-1659. doi: 10.1002/humu.23341. Epub 2017 Oct 6. Hum Mutat. 2017. PMID: 28940506 Review.
-
Characterization of the C584R variant in the mtDNA depletion syndrome gene FBXL4, reveals a novel role for FBXL4 as a regulator of mitochondrial fusion.Biochim Biophys Acta Mol Basis Dis. 2019 Nov 1;1865(11):165536. doi: 10.1016/j.bbadis.2019.165536. Epub 2019 Aug 20. Biochim Biophys Acta Mol Basis Dis. 2019. PMID: 31442532
-
Disorders associated with depletion of mitochondrial DNA.Brain Pathol. 1992 Apr;2(2):141-7. doi: 10.1111/j.1750-3639.1992.tb00682.x. Brain Pathol. 1992. PMID: 1341955 Review.
Cited by
-
Dual-localized PPTC7 limits mitophagy through proximal and dynamic interactions with BNIP3 and NIX.Life Sci Alliance. 2024 Jul 11;7(9):e202402765. doi: 10.26508/lsa.202402765. Print 2024 Sep. Life Sci Alliance. 2024. PMID: 38991726 Free PMC article.
-
Dynamic features of human mitochondrial DNA maintenance and transcription.Front Cell Dev Biol. 2022 Sep 7;10:984245. doi: 10.3389/fcell.2022.984245. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36158192 Free PMC article. Review.
-
Mitochondrial Dysfunction in Peripheral Blood Mononuclear Cells as Novel Diagnostic Tools for Non-Alcoholic Fatty Liver Disease: Visualizing Relationships with Known and Potential Disease Biomarkers.Diagnostics (Basel). 2023 Jul 13;13(14):2363. doi: 10.3390/diagnostics13142363. Diagnostics (Basel). 2023. PMID: 37510108 Free PMC article.
-
Age-related mitochondrial DNA depletion and the impact on pancreatic Beta cell function.PLoS One. 2014 Dec 22;9(12):e115433. doi: 10.1371/journal.pone.0115433. eCollection 2014. PLoS One. 2014. PMID: 25532126 Free PMC article.
-
FBXL4 suppresses mitophagy by restricting the accumulation of NIX and BNIP3 mitophagy receptors.EMBO J. 2023 Jul 3;42(13):e112767. doi: 10.15252/embj.2022112767. Epub 2023 May 10. EMBO J. 2023. PMID: 37161784 Free PMC article.
References
-
- Debray F.G., Lambert M., Mitchell G.A. Disorders of mitochondrial function. Curr. Opin. Pediatr. 2008;20:471–482. - PubMed
-
- Koopman W.J.H., Willems P.H.G.M., Smeitink J.A.M. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. - PubMed
-
- Thorburn D.R. Mitochondrial disorders: prevalence, myths and advances. J. Inherit. Metab. Dis. 2004;27:349–362. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- CA125123/CA/NCI NIH HHS/United States
- MR/K000608/1/MRC_/Medical Research Council/United Kingdom
- R01 GM098387/GM/NIGMS NIH HHS/United States
- 096919/Z/11/Z/WT_/Wellcome Trust/United Kingdom
- P30 CA125123/CA/NCI NIH HHS/United States
- P30 AI036211/AI/NIAID NIH HHS/United States
- S10 RR024574/RR/NCRR NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- RR024574/RR/NCRR NIH HHS/United States
- AI036211/AI/NIAID NIH HHS/United States
- G0601943/MRC_/Medical Research Council/United Kingdom
- G0800674/MRC_/Medical Research Council/United Kingdom
- 096919/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
