

Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients
- PMID: 22742934
- PMCID: PMC3500692
- DOI: 10.1016/j.nmd.2012.05.001
Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients
Abstract
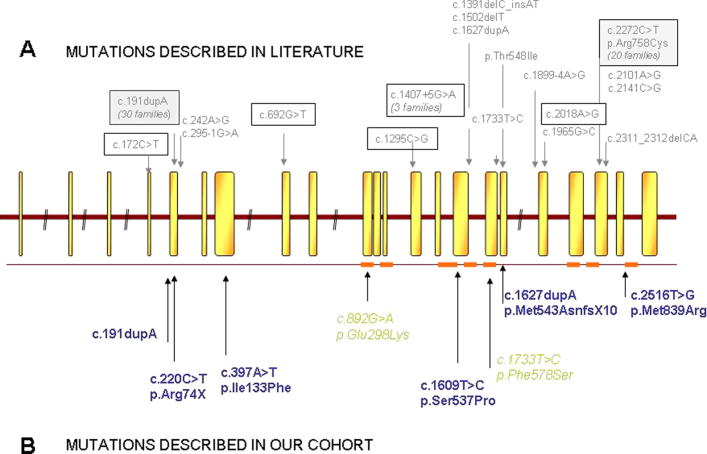
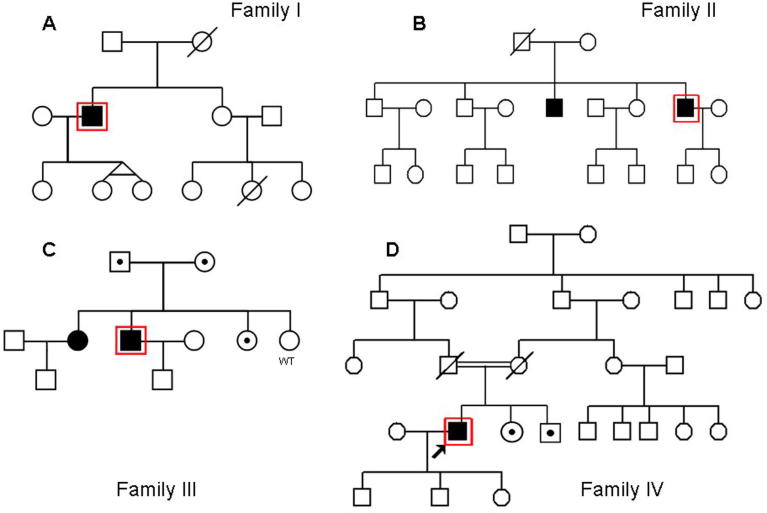
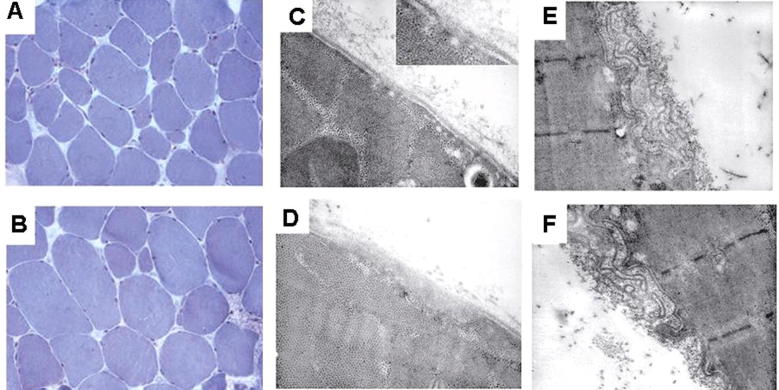
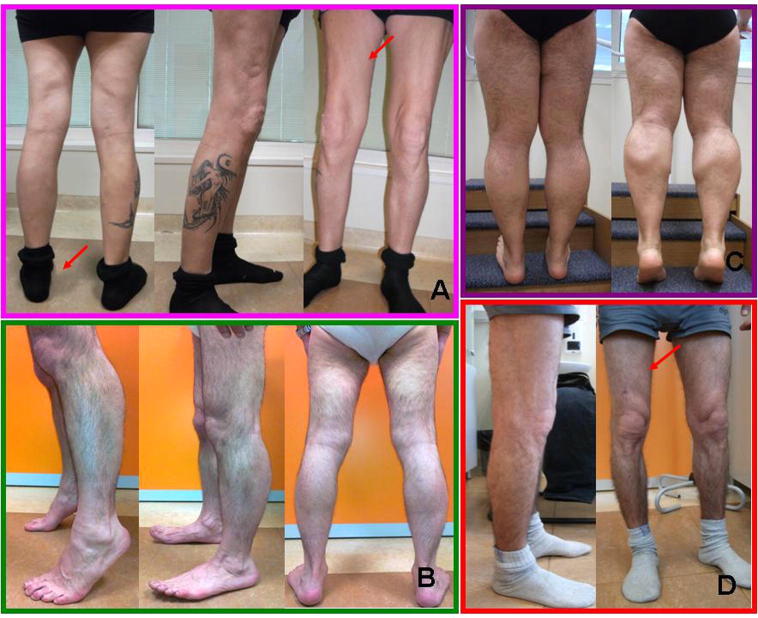
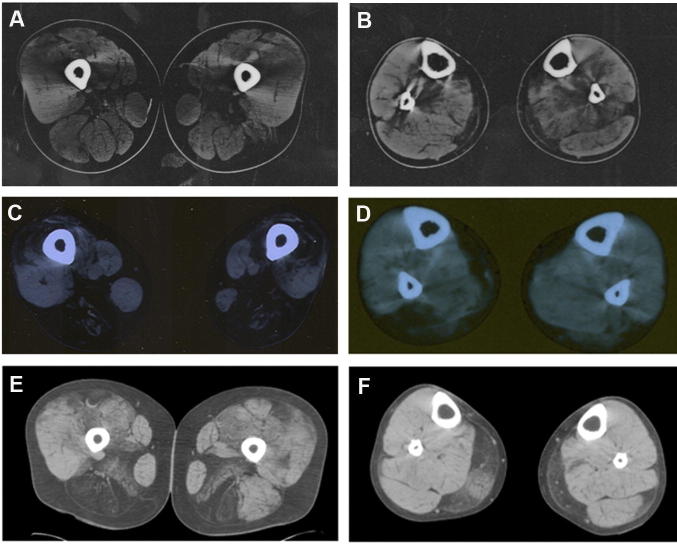
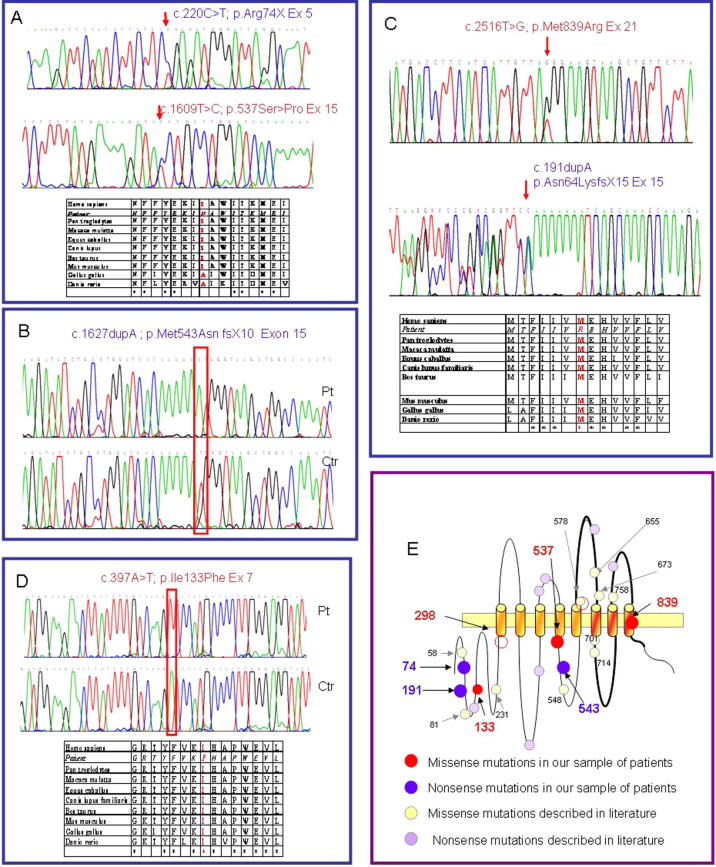
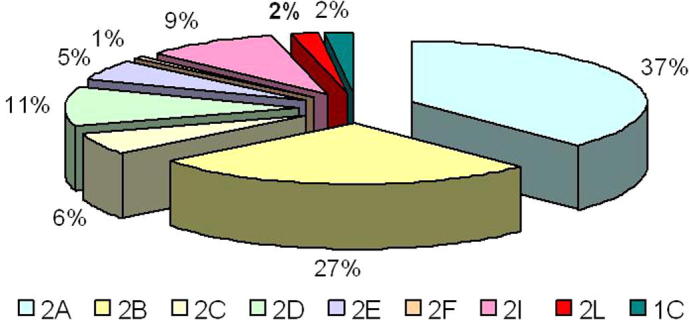
Limb-girdle muscular dystrophy (LGMD) 2L, caused by mutations in the anoctamin 5 (ANO5) gene, is the third most common LGMD in Northern and Central Europe, where the c.191dupA mutation causes the majority of cases. We evaluated data from 228 Italian LGMD patients to determine the prevalence of LGMD2L and the c.191dupA mutation, and to describe the clinical, muscle biopsy, and magnetic resonance imaging findings in these patients. Forty-three patients who lacked molecular diagnosis were studied for ANO5 mutations, and four novel mutations were found in three probands. Only one proband carried the c.191dupA mutation, which was compound heterozygous with c.2516T>G. Two probands were homozygous for the c.1627dupA and c.397A>T mutations, respectively, while a fourth proband had a compound heterozygous status (c.220C>T and c.1609T>C). Therefore occurrence and molecular epidemiology of LGMD2L in this Italian cohort differed from those observed in other European countries. ANO5 mutations accounted for ∼2% of our sample. Affected patients exhibited benign progression with variable onset and an absence of cardiac and respiratory impairment; muscle biopsy generally showed mild signs, except when performed on the quadriceps muscles; MRI showed predominant involvement of the posterior thigh. Overall these common clinical, morphological and imaging findings could be useful in differential diagnosis.
Copyright © 2012 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy.Brain. 2011 Jan;134(Pt 1):171-182. doi: 10.1093/brain/awq294. Brain. 2011. PMID: 21186264 Free PMC article.
-
Novel ANO5 mutations causing hyper-CK-emia, limb girdle muscular weakness and Miyoshi type of muscular dystrophy.Muscle Nerve. 2012 May;45(5):740-2. doi: 10.1002/mus.23281. Muscle Nerve. 2012. PMID: 22499103
-
[Muscular dystrophy due to mutations in anoctamin 5: clinical and molecular genetic findings].Nervenarzt. 2011 Dec;82(12):1596-603. doi: 10.1007/s00115-011-3325-4. Nervenarzt. 2011. PMID: 21739273 German.
-
Anoctamin 5 (ANO5) Muscle Disorders: A Narrative Review.Genes (Basel). 2022 Sep 27;13(10):1736. doi: 10.3390/genes13101736. Genes (Basel). 2022. PMID: 36292621 Free PMC article. Review.
-
Limb-girdle muscular dystrophy due to GMPPB mutations: A case report and comprehensive literature review.Bosn J Basic Med Sci. 2020 May 1;20(2):275-280. doi: 10.17305/bjbms.2019.3992. Bosn J Basic Med Sci. 2020. PMID: 30684953 Free PMC article. Review.
Cited by
-
The Popeye Domain Containing Genes and their Function in Striated Muscle.J Cardiovasc Dev Dis. 2016 Jun 15;3(2):22. doi: 10.3390/jcdd3020022. J Cardiovasc Dev Dis. 2016. PMID: 27347491 Free PMC article.
-
Limb girdle muscular dystrophy type 2L presenting as necrotizing myopathy.Acta Myol. 2014 May;33(1):19-21. Acta Myol. 2014. PMID: 24843231 Free PMC article.
-
Deficiency of Anoctamin 5/TMEM16E causes nuclear positioning defect and impairs Ca2+ signaling of differentiated C2C12 myotubes.Korean J Physiol Pharmacol. 2019 Nov;23(6):539-547. doi: 10.4196/kjpp.2019.23.6.539. Epub 2019 Oct 24. Korean J Physiol Pharmacol. 2019. PMID: 31680776 Free PMC article.
-
Clinical and genetic features of anoctaminopathy in Saudi Arabia.Neurosciences (Riyadh). 2015 Apr;20(2):173-7. doi: 10.17712/nsj.2015.2.20140547. Neurosciences (Riyadh). 2015. PMID: 25864073 Free PMC article.
-
ANO5-related muscle diseases: From clinics and genetics to pathology and research strategies.Genes Dis. 2022 Feb 14;9(6):1506-1520. doi: 10.1016/j.gendis.2022.01.001. eCollection 2022 Nov. Genes Dis. 2022. PMID: 36157496 Free PMC article. Review.
References
-
- Kunzelmann K., Tian Y., Martins J.R. Anoctamins. Pflugers Arch. 2011 Aug;462(2):195–208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
