

Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome
- PMID: 22577224
- PMCID: PMC4771841
- DOI: 10.1136/jmedgenet-2011-100686
Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome
Abstract
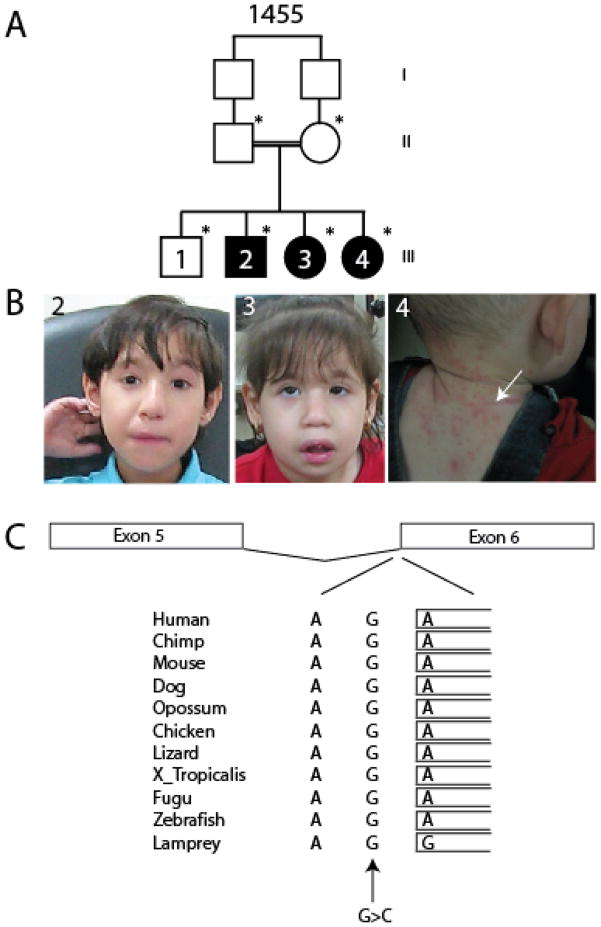
Background: Dubowitz syndrome (DS) is an autosomal recessive disorder characterized by the constellation of mild microcephaly, growth and mental retardation, eczema and peculiar facies. Over 140 cases have been reported, but the genetic basis is not understood.
Methods: We enrolled a multiplex consanguineous family from the United Arab Emirates with many of the key clinical features of DS as reported in previous series. The family was analyzed by whole exome sequencing. RNA splicing was evaluated with reverse-transcriptase PCR, immunostaining and western blotting was performed with specific antibodies, and site-specific cytosine-5-methylation was studied with bisulfite sequencing.
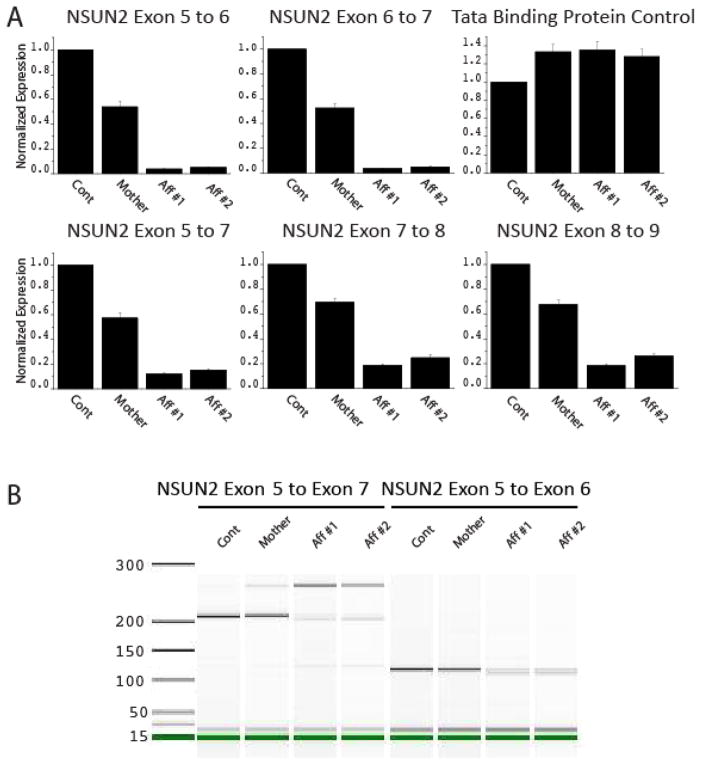
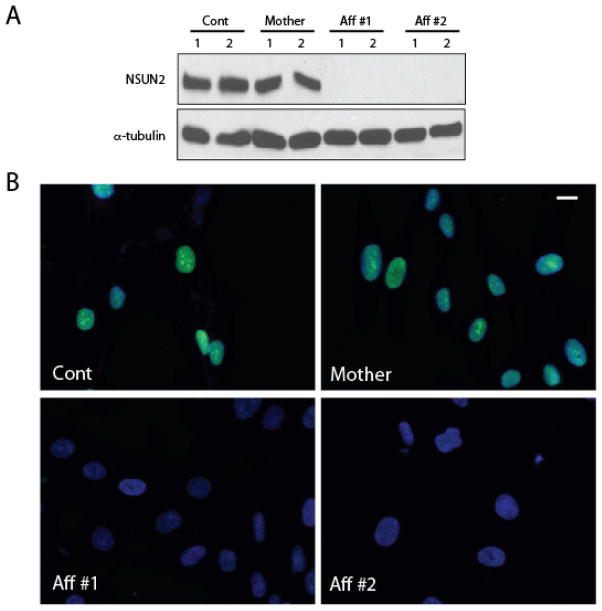
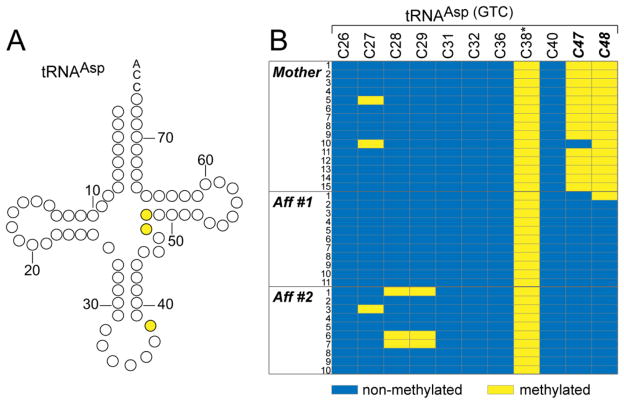
Results: We identified a homozygous splice mutation in the NSUN2 gene, encoding a conserved RNA methyltransferase. The mutation abolished the canonical splice acceptor site of exon 6, leading to use of a cryptic splice donor within an AluY and subsequent mRNA instability. Patient cells lacked NSUN2 protein and there was resultant loss of site-specific 5-cytosine methylation of the tRNA(Asp GTC) at C47 and C48, known NSUN2 targets.
Conclusion: Our findings establish NSUN2 as the first causal gene with relationship to the DS spectrum phenotype. NSUN2 has been implicated in Myc-induced cell proliferation and mitotic spindle stability, which might help explain the varied clinical presentation in DS that can include chromosomal instability and immunological defects.
Figures
Comment in
-
Targeting branch sites of new exons?J Med Genet. 2012 Jul;49(7):480. doi: 10.1136/jmedgenet-2012-101055. Epub 2012 Jun 22. J Med Genet. 2012. PMID: 22730556 Free PMC article. No abstract available.
Similar articles
-
A novel variant in NSUN2 causes intellectual disability in a Chinese family.BMC Med Genomics. 2024 Apr 20;17(1):95. doi: 10.1186/s12920-024-01883-x. BMC Med Genomics. 2024. PMID: 38643142 Free PMC article.
-
Further delineation of autosomal recessive intellectual disability syndrome caused by homozygous variant of the NSUN2 gene in a chinese pedigree.Mol Genet Genomic Med. 2020 Dec;8(12):e1518. doi: 10.1002/mgg3.1518. Epub 2020 Oct 1. Mol Genet Genomic Med. 2020. PMID: 33002343 Free PMC article.
-
Alternative genomic diagnoses for individuals with a clinical diagnosis of Dubowitz syndrome.Am J Med Genet A. 2021 Jan;185(1):119-133. doi: 10.1002/ajmg.a.61926. Epub 2020 Oct 24. Am J Med Genet A. 2021. PMID: 33098347 Free PMC article.
-
SETD5 loss-of-function mutation as a likely cause of a familial syndromic intellectual disability with variable phenotypic expression.Am J Med Genet A. 2016 Sep;170(9):2322-7. doi: 10.1002/ajmg.a.37832. Epub 2016 Jul 4. Am J Med Genet A. 2016. PMID: 27375234 Review.
-
A recessive syndrome of intellectual disability, moderate overgrowth, and renal dysplasia predisposing to Wilms tumor is caused by a mutation in FIBP gene.Am J Med Genet A. 2016 Aug;170(8):2111-8. doi: 10.1002/ajmg.a.37741. Epub 2016 May 17. Am J Med Genet A. 2016. PMID: 27183861 Review.
Cited by
-
The RNA Methyltransferase NSUN2 and Its Potential Roles in Cancer.Cells. 2020 Jul 22;9(8):1758. doi: 10.3390/cells9081758. Cells. 2020. PMID: 32708015 Free PMC article. Review.
-
PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly.Hum Genet. 2019 Mar;138(3):231-239. doi: 10.1007/s00439-019-01980-3. Epub 2019 Feb 18. Hum Genet. 2019. PMID: 30778726 Free PMC article.
-
Neuronal Nsun2 deficiency produces tRNA epitranscriptomic alterations and proteomic shifts impacting synaptic signaling and behavior.Nat Commun. 2021 Aug 13;12(1):4913. doi: 10.1038/s41467-021-24969-x. Nat Commun. 2021. PMID: 34389722 Free PMC article.
-
RNA 5-methylcytosine modification and its emerging role as an epitranscriptomic mark.RNA Biol. 2021 Oct 15;18(sup1):117-127. doi: 10.1080/15476286.2021.1950993. Epub 2021 Jul 21. RNA Biol. 2021. PMID: 34288807 Free PMC article. Review.
-
Deciphering the epitranscriptome: A green perspective.J Integr Plant Biol. 2016 Oct;58(10):822-835. doi: 10.1111/jipb.12483. Epub 2016 Jun 20. J Integr Plant Biol. 2016. PMID: 27172004 Free PMC article. Review.
References
-
- Tsukahara M, Opitz JM. Dubowitz syndrome: review of 141 cases including 36 previously unreported patients. Am J Med Genet. 1996;63(1):277–89. - PubMed
-
- Dentici ML, Mingarelli R, Dallapiccola B. The difficult nosology of blepharophimosis-mental retardation syndromes: report on two siblings. Am J Med Genet A. 2011;155A(3):459–65. - PubMed
-
- Al-Nemri AR, Kilani RA, Salih MA, Al-Ajlan AA. Embryonal rhabdomyosarcoma and chromosomal breakage in a newborn infant with possible Dubowitz syndrome. Am J Med Genet. 2000;92(2):107–10. - PubMed
-
- Clayton-Smith J, O’Sullivan J, Daly S, Bhaskar S, Day R, Anderson B, Voss AK, Thomas T, Biesecker LG, Smith P, Fryer A, Chandler KE, Kerr B, Tassabehji M, Lynch SA, Krajewska-Walasek M, McKee S, Smith J, Sweeney E, Mansour S, Mohammed S, Donnai D, Black G. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89(5):675–81. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- HHMI/Howard Hughes Medical Institute/United States
- U54HG003067/HG/NHGRI NIH HHS/United States
- P01HD070494/HD/NICHD NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- 079249/Wellcome Trust/United Kingdom
- P30NS047101/NS/NINDS NIH HHS/United States
- R01NS048453/NS/NINDS NIH HHS/United States
- P30 NS047101/NS/NINDS NIH HHS/United States
- R01 NS048453/NS/NINDS NIH HHS/United States
- G0801904/MRC_/Medical Research Council/United Kingdom
- R01 NS052455/NS/NINDS NIH HHS/United States
- P01 HD070494/HD/NICHD NIH HHS/United States
- G0800784/MRC_/Medical Research Council/United Kingdom
- R01NS052455/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous