

Mutations in KAT6B, encoding a histone acetyltransferase, cause Genitopatellar syndrome
- PMID: 22265014
- PMCID: PMC3276659
- DOI: 10.1016/j.ajhg.2011.11.023
Mutations in KAT6B, encoding a histone acetyltransferase, cause Genitopatellar syndrome
Abstract
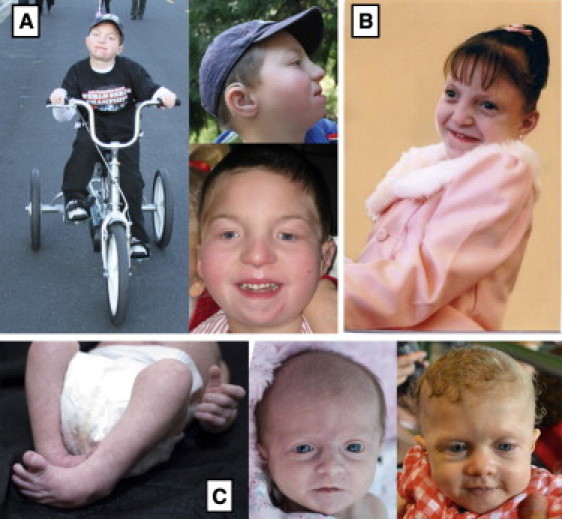
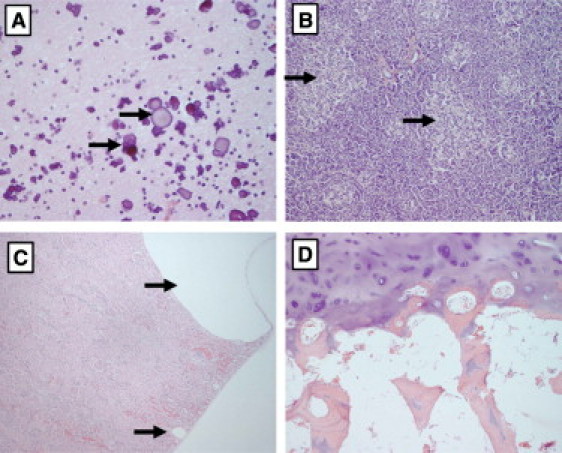
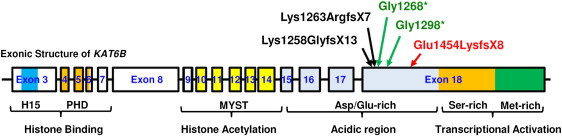
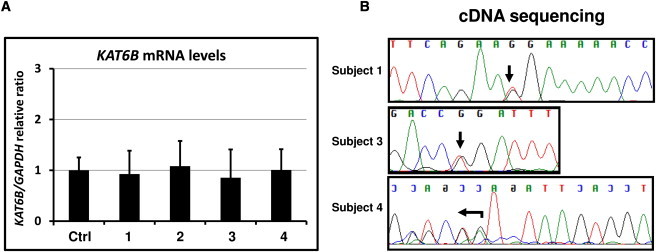
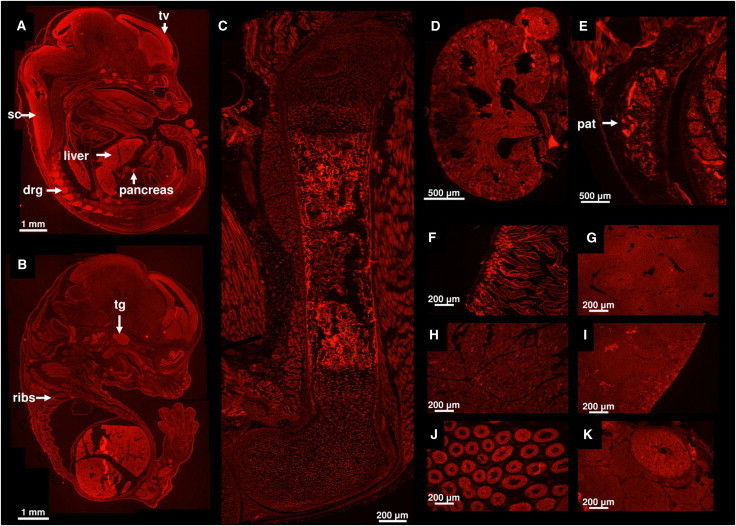
Genitopatellar syndrome (GPS) is a skeletal dysplasia with cerebral and genital anomalies for which the molecular basis has not yet been determined. By exome sequencing, we found de novo heterozygous truncating mutations in KAT6B (lysine acetyltransferase 6B, formerly known as MYST4 and MORF) in three subjects; then by Sanger sequencing of KAT6B, we found similar mutations in three additional subjects. The mutant transcripts do not undergo nonsense-mediated decay in cells from subjects with GPS. In addition, human pathological analyses and mouse expression studies point to systemic roles of KAT6B in controlling organismal growth and development. Myst4 (the mouse orthologous gene) is expressed in mouse tissues corresponding to those affected by GPS. Phenotypic differences and similarities between GPS, the Say-Barber-Biesecker variant of Ohdo syndrome (caused by different mutations of KAT6B), and Rubinstein-Taybi syndrome (caused by mutations in other histone acetyltransferases) are discussed. Together, the data support an epigenetic dysregulation of the limb, brain, and genital developmental programs.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms.Hum Mutat. 2012 Nov;33(11):1520-5. doi: 10.1002/humu.22141. Epub 2012 Jul 12. Hum Mutat. 2012. PMID: 22715153 Free PMC article. Review.
-
A patient showing features of both SBBYSS and GPS supports the concept of a KAT6B-related disease spectrum, with mutations in mid-exon 18 possibly leading to combined phenotypes.Eur J Med Genet. 2015 Oct;58(10):550-5. doi: 10.1016/j.ejmg.2015.09.004. Epub 2015 Sep 11. Eur J Med Genet. 2015. PMID: 26370006
-
De novo mutations of the gene encoding the histone acetyltransferase KAT6B cause Genitopatellar syndrome.Am J Hum Genet. 2012 Feb 10;90(2):290-4. doi: 10.1016/j.ajhg.2011.11.024. Epub 2012 Jan 19. Am J Hum Genet. 2012. PMID: 22265017 Free PMC article.
-
A novel truncating variant within exon 7 of KAT6B associated with features of both Say-Barber-Bieseker-Young-Simpson syndrome and genitopatellar syndrome: Further evidence of a continuum in the clinical spectrum of KAT6B-related disorders.Am J Med Genet A. 2018 Feb;176(2):455-459. doi: 10.1002/ajmg.a.38571. Epub 2017 Dec 11. Am J Med Genet A. 2018. PMID: 29226580
-
Further delineation of the clinical spectrum of KAT6B disorders and allelic series of pathogenic variants.Genet Med. 2020 Aug;22(8):1338-1347. doi: 10.1038/s41436-020-0811-8. Epub 2020 May 19. Genet Med. 2020. PMID: 32424177 Free PMC article. Review.
Cited by
-
Chromatin modifiers and histone modifications in bone formation, regeneration, and therapeutic intervention for bone-related disease.Bone. 2015 Dec;81:739-745. doi: 10.1016/j.bone.2015.03.011. Epub 2015 Mar 31. Bone. 2015. PMID: 25836763 Free PMC article. Review.
-
Genetics of the patella.Eur J Hum Genet. 2019 May;27(5):671-680. doi: 10.1038/s41431-018-0329-6. Epub 2019 Jan 21. Eur J Hum Genet. 2019. PMID: 30664715 Free PMC article. Review.
-
Clinical features and underlying mechanisms of KAT6B disease in a Chinese boy.Mol Genet Genomic Med. 2023 Sep;11(9):e2202. doi: 10.1002/mgg3.2202. Epub 2023 Jun 8. Mol Genet Genomic Med. 2023. PMID: 37288707 Free PMC article.
-
A Molecular Perspective on Sirtuin Activity.Int J Mol Sci. 2020 Nov 15;21(22):8609. doi: 10.3390/ijms21228609. Int J Mol Sci. 2020. PMID: 33203121 Free PMC article. Review.
-
Recent advancements in understanding the role of epigenetics in the auditory system.Gene. 2020 Nov 30;761:144996. doi: 10.1016/j.gene.2020.144996. Epub 2020 Jul 29. Gene. 2020. PMID: 32738421 Free PMC article. Review.
References
-
- Abdul-Rahman O.A., La T.H., Kwan A., Schlaubitz S., Barsh G.S., Enns G.M., Hudgins L. Genitopatellar syndrome: Expanding the phenotype and excluding mutations in LMX1B and TBX4. Am. J. Med. Genet. A. 2006;140:1567–1572. - PubMed
-
- Bergmann C., Spranger S., Javaher P., Ptok M. Genitopatellar syndrome, sensorineural hearing loss, and cleft palate. Oral Maxillofac Surg. 2011;15:103–106. - PubMed
-
- Brugha R., Kinali M., Aminu K., Bridges N., Holder S.E. Genitopatellar syndrome: A further case. Clin. Dysmorphol. 2011;20:163–165. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
