

Natural history of infantile G(M2) gangliosidosis
- PMID: 22025593
- PMCID: PMC3208966
- DOI: 10.1542/peds.2011-0078
Natural history of infantile G(M2) gangliosidosis
Abstract
Objective: G(M2) gangliosidoses are caused by an inherited deficiency of lysosomal β-hexosaminidase and result in ganglioside accumulation in the brain. Onset during infancy leads to rapid neurodegeneration and death before 4 years of age. We set out to quantify the rate of functional decline in infantile G(M2) gangliosidosis on the basis of patient surveys and a comprehensive review of existing literature.
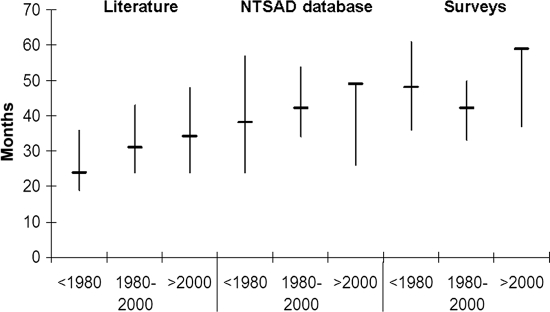
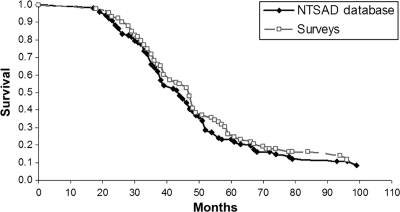
Methods: Patients with infantile G(M2) gangliosidosis (N = 237) were surveyed via questionnaire by the National Tay Sachs & Allied Diseases Association (NTSAD). These data were supplemented by survival data from the NTSAD database and a literature survey. Detailed retrospective surveys from 97 patients were available. Five patients who had received hematopoietic stem cell transplantation were evaluated separately. The mortality rate of the remaining 92 patients was comparable to that of the 103 patients from the NTSAD database and 121 patients reported in the literature.
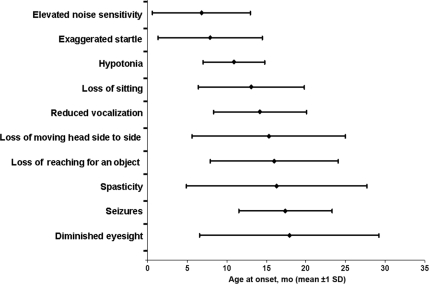
Results: Common symptoms at onset were developmental arrest (83%), startling (65%), and hypotonia (60%). All 55 patients who had learned to sit without support lost that ability within 1 year. Individual functional measures correlated with each other but not with survival. Gastric tube placement was associated with prolonged survival. Tay Sachs and Sandhoff variants did not differ. Hematopoietic stem cell transplantation was not associated with prolonged survival.
Conclusions: We studied the timing of regression in 97 cases of infantile G(M2) gangliosidosis and conclude that clinical disease progression does not correlate with survival, likely because of the impact of improved supportive care over time. However, functional measures are quantifiable and can inform power calculations and study design of future interventions.
Figures
Similar articles
-
Infantile gangliosidoses: Mapping a timeline of clinical changes.Mol Genet Metab. 2017 Jun;121(2):170-179. doi: 10.1016/j.ymgme.2017.04.011. Epub 2017 Apr 29. Mol Genet Metab. 2017. PMID: 28476546 Free PMC article. Clinical Trial.
-
The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported.Pediatrics. 2006 Nov;118(5):e1550-62. doi: 10.1542/peds.2006-0588. Epub 2006 Oct 2. Pediatrics. 2006. PMID: 17015493 Free PMC article. Review.
-
Distinct progression patterns of brain disease in infantile and juvenile gangliosidoses: Volumetric quantitative MRI study.Mol Genet Metab. 2018 Feb;123(2):97-104. doi: 10.1016/j.ymgme.2017.12.432. Epub 2017 Dec 20. Mol Genet Metab. 2018. PMID: 29352662 Free PMC article.
-
Clinical outcome assessments of disease burden and progression in late-onset GM2 gangliosidoses.Mol Genet Metab. 2024 Jul;142(3):108512. doi: 10.1016/j.ymgme.2024.108512. Epub 2024 Jun 6. Mol Genet Metab. 2024. PMID: 38870773
-
[Molecular pathogenesis and therapeutic approach of GM2 gangliosidosis].Yakugaku Zasshi. 2013;133(2):269-74. doi: 10.1248/yakushi.12-00199. Yakugaku Zasshi. 2013. PMID: 23370522 Review. Japanese.
Cited by
-
Lysosomal Storage Disease-Associated Neuropathy: Targeting Stable Nucleic Acid Lipid Particle (SNALP)-Formulated siRNAs to the Brain as a Therapeutic Approach.Int J Mol Sci. 2020 Aug 10;21(16):5732. doi: 10.3390/ijms21165732. Int J Mol Sci. 2020. PMID: 32785133 Free PMC article. Review.
-
Characterization of a phenotypically severe animal model for human AB-Variant GM2 gangliosidosis.Front Mol Neurosci. 2023 Nov 30;16:1242814. doi: 10.3389/fnmol.2023.1242814. eCollection 2023. Front Mol Neurosci. 2023. PMID: 38098938 Free PMC article.
-
Clinical and Laboratory Profile of Gangliosidosis from Southern Part of India.J Pediatr Genet. 2020 Oct 19;11(1):34-41. doi: 10.1055/s-0040-1718726. eCollection 2022 Mar. J Pediatr Genet. 2020. PMID: 35186388 Free PMC article.
-
sp2-Iminosugars targeting human lysosomal β-hexosaminidase as pharmacological chaperone candidates for late-onset Tay-Sachs disease.J Enzyme Inhib Med Chem. 2022 Dec;37(1):1364-1374. doi: 10.1080/14756366.2022.2073444. J Enzyme Inhib Med Chem. 2022. PMID: 35575117 Free PMC article.
-
Three novel mutations in Iranian patients with Tay-Sachs disease.Iran Biomed J. 2014;18(2):114-9. doi: 10.6091/ibj.1137.2013. Iran Biomed J. 2014. PMID: 24518553 Free PMC article.
References
-
- Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K, Suzuki K. The GM2 gangliosidoses. In: Scriver C, Beaudet A, Sly W, Valle D. eds. Metabolic and Molecular Bases of Inherited Diseases. New York, NY: McGraw-Hill; 2001:3827–3876
-
- Maegawa GH, Stockley T, Tropak M, et al. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported [published correction appears in Pediatrics. 2007;120(4):936]. Pediatrics. 2006;118(5). Available at: www.pediatrics.org/cgi/content/full/118/5/e1550 - PMC - PubMed
-
- Neudorfer O, Pastores GM, Zeng BJ, et al. Late-onset Tay-Sachs disease: phenotypic characterization and genotypic correlations in 21 affected patients. Genet Med. 2005;7(2):119–123 - PubMed
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–254 - PubMed
-
- Broekman ML, Tierney LA, Benn C, Chawla P, Cha JH, Sena-Esteves M. Mechanisms of distribution of mouse beta-galactosidase in the adult GM1-gangliosidosis brain. Gene Ther. 2009;16(2):303–308 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
