

Legius syndrome in fourteen families
- PMID: 21089071
- PMCID: PMC3038325
- DOI: 10.1002/humu.21404
Legius syndrome in fourteen families
Abstract
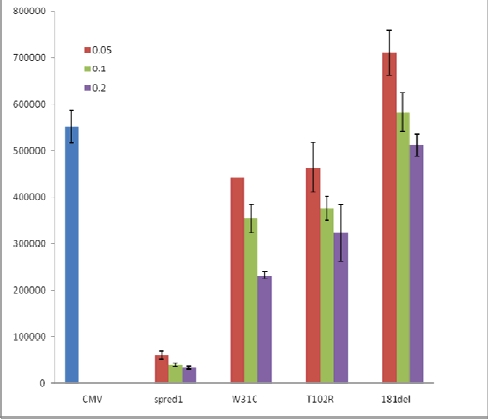
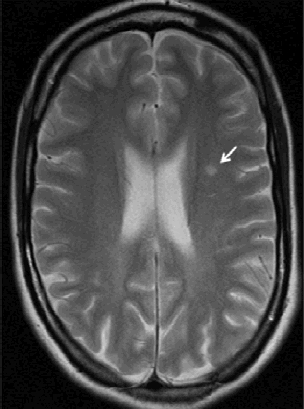
Legius syndrome presents as an autosomal dominant condition characterized by café-au-lait macules with or without freckling and sometimes a Noonan-like appearance and/or learning difficulties. It is caused by germline loss-of-function SPRED1 mutations and is a member of the RAS-MAPK pathway syndromes. Most mutations result in a truncated protein and only a few inactivating missense mutations have been reported. Since only a limited number of patients has been reported up until now, the full clinical and mutational spectrum is still unknown. We report mutation data and clinical details in fourteen new families with Legius syndrome. Six novel germline mutations are described. The Trp31Cys mutation is a new pathogenic SPRED1 missense mutation. Clinical details in the 14 families confirmed the absence of neurofibromas, and Lisch nodules, and the absence of a high prevalence of central nervous system tumors. We report white matter T2 hyperintensities on brain MRI scans in 2 patients and a potential association between postaxial polydactyly and Legius syndrome.
© 2010 Wiley-Liss, Inc.
Figures
Similar articles
-
Novel causative variants in Legius syndrome: SPRED1 Genotype spectrum expansion.Am J Med Genet A. 2024 Dec;194(12):e63824. doi: 10.1002/ajmg.a.63824. Epub 2024 Jul 19. Am J Med Genet A. 2024. PMID: 39031930
-
Interaction between a Domain of the Negative Regulator of the Ras-ERK Pathway, SPRED1 Protein, and the GTPase-activating Protein-related Domain of Neurofibromin Is Implicated in Legius Syndrome and Neurofibromatosis Type 1.J Biol Chem. 2016 Feb 12;291(7):3124-34. doi: 10.1074/jbc.M115.703710. Epub 2015 Dec 3. J Biol Chem. 2016. PMID: 26635368 Free PMC article.
-
Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome.JAMA. 2009 Nov 18;302(19):2111-8. doi: 10.1001/jama.2009.1663. JAMA. 2009. PMID: 19920235
-
Review and update of SPRED1 mutations causing Legius syndrome.Hum Mutat. 2012 Nov;33(11):1538-46. doi: 10.1002/humu.22152. Epub 2012 Aug 1. Hum Mutat. 2012. PMID: 22753041 Review.
-
Legius syndrome, an Update. Molecular pathology of mutations in SPRED1.Keio J Med. 2013;62(4):107-12. doi: 10.2302/kjm.2013-0002-re. Epub 2013 Dec 10. Keio J Med. 2013. PMID: 24334617 Review.
Cited by
-
Autoimmune Thyroid Disease in Specific Genetic Syndromes in Childhood and Adolescence.Front Endocrinol (Lausanne). 2020 Aug 19;11:543. doi: 10.3389/fendo.2020.00543. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 32973676 Free PMC article. Review.
-
The SPRED1 Variants Repository for Legius Syndrome.G3 (Bethesda). 2011 Nov;1(6):451-6. doi: 10.1534/g3.111.000687. Epub 2011 Nov 1. G3 (Bethesda). 2011. PMID: 22384355 Free PMC article.
-
The NF1 gene in tumor syndromes and melanoma.Lab Invest. 2017 Feb;97(2):146-157. doi: 10.1038/labinvest.2016.142. Epub 2017 Jan 9. Lab Invest. 2017. PMID: 28067895 Free PMC article. Review.
-
MEK inhibition ameliorates social behavior phenotypes in a Spred1 knockout mouse model for RASopathy disorders.Mol Autism. 2021 Jul 26;12(1):53. doi: 10.1186/s13229-021-00458-2. Mol Autism. 2021. PMID: 34311771 Free PMC article.
-
Non-Mammalian Models for Understanding Neurological Defects in RASopathies.Biomedicines. 2024 Apr 10;12(4):841. doi: 10.3390/biomedicines12040841. Biomedicines. 2024. PMID: 38672195 Free PMC article. Review.
References
-
- Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283–285. - PubMed
-
- Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns JP, Cools J, Marynen P, Thomas G, Yoshimura A, Legius E. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39:1120–1126. - PubMed
-
- Denayer E, de Ravel T, Legius E. Clinical and molecular aspects of RAS related disorders. J Med Genet. 2008;45:695–703. - PubMed
-
- Gill DS, Hyman SL, Steinberg A, North KN. Age-related findings on MRI in neurofibromatosis type 1. Pediatr Radiol. 2006;36:1048–1056. - PubMed
-
- Kehrer-Sawatzki H, Cooper DN. Mosaicism in sporadic neurofibromatosis type 1: variations on a theme common to other hereditary cancer syndromes? J Med Genet. 2008;45:622–631. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
