

Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology
- PMID: 20934517
- PMCID: PMC3040982
- DOI: 10.1016/j.semcancer.2010.10.002
Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology
Abstract
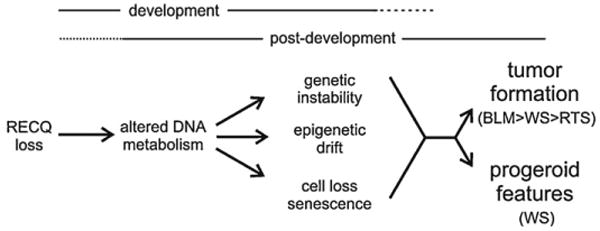
Helicases use the energy of ATP hydrolysis to separate double-stranded nucleic acids to facilitate essential processes such as replication, recombination, transcription and repair. This article focuses on the human RECQ helicase gene and protein family. Loss of function of three different members has been shown to cause Bloom syndrome (BS), Werner syndrome (WS) and Rothmund-Thomson syndrome (RTS). This article outlines clinical and cellular features of these cancer predisposition syndromes, and discusses their pathogenesis in light of our understanding of RECQ helicase biochemical activities and in vivo functions. I also discuss the emerging role for RECQ helicases as predictors of disease risk and the response to therapy.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Conflict of interest statement
R.J.M., Jr. has no relevant conflicts of interest to declare.
Figures
Similar articles
-
Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells.Cancer Lett. 2018 Jan 28;413:1-10. doi: 10.1016/j.canlet.2017.10.021. Epub 2017 Nov 7. Cancer Lett. 2018. PMID: 29080750 Review.
-
Human RecQL4 as a Novel Molecular Target for Cancer Therapy.Cytogenet Genome Res. 2021;161(6-7):305-327. doi: 10.1159/000516568. Epub 2021 Sep 2. Cytogenet Genome Res. 2021. PMID: 34474412 Review.
-
Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance.Trends Biochem Sci. 2008 Dec;33(12):609-20. doi: 10.1016/j.tibs.2008.09.003. Epub 2008 Oct 14. Trends Biochem Sci. 2008. PMID: 18926708 Free PMC article. Review.
-
Premature aging and predisposition to cancers caused by mutations in RecQ family helicases.Ann N Y Acad Sci. 2001 Apr;928:121-31. doi: 10.1111/j.1749-6632.2001.tb05642.x. Ann N Y Acad Sci. 2001. PMID: 11795503 Review.
-
Bloom's syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases.Ageing Res Rev. 2017 Jan;33:36-51. doi: 10.1016/j.arr.2016.05.010. Epub 2016 May 26. Ageing Res Rev. 2017. PMID: 27238185 Free PMC article. Review.
Cited by
-
DNA helicases involved in DNA repair and their roles in cancer.Nat Rev Cancer. 2013 Aug;13(8):542-58. doi: 10.1038/nrc3560. Epub 2013 Jul 11. Nat Rev Cancer. 2013. PMID: 23842644 Free PMC article. Review.
-
Loss of the bloom syndrome helicase increases DNA ligase 4-independent genome rearrangements and tumorigenesis in aging Drosophila.Genome Biol. 2011 Dec 19;12(12):R121. doi: 10.1186/gb-2011-12-12-r121. Genome Biol. 2011. PMID: 22183041 Free PMC article.
-
Replication stress induces specific enrichment of RECQ1 at common fragile sites FRA3B and FRA16D.Mol Cancer. 2013 Apr 22;12(1):29. doi: 10.1186/1476-4598-12-29. Mol Cancer. 2013. PMID: 23601052 Free PMC article.
-
Low expression of RecQ-like helicase 5 is associated with poor prognosis in patients with gastric cancer.Oncol Lett. 2020 Jan;19(1):985-991. doi: 10.3892/ol.2019.11137. Epub 2019 Nov 21. Oncol Lett. 2020. PMID: 31897211 Free PMC article.
-
Resolution by unassisted Top3 points to template switch recombination intermediates during DNA replication.J Biol Chem. 2013 Nov 15;288(46):33193-204. doi: 10.1074/jbc.M113.496133. Epub 2013 Oct 7. J Biol Chem. 2013. PMID: 24100144 Free PMC article.
References
-
- Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76(1):23–50. - PubMed
-
- Vindigni A, Marino F, Gileadi O. Probing the structural basis of RecQ helicase function. Biophys Chem. 2010;149:67–77. - PubMed
-
- Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs. Am J Dis Child. 1954;88:754–8. - PubMed
-
- German J. Bloom syndrome: a Mendelian prototype of somatic mutational disease. Medicine. 1993;72:393–406. - PubMed
-
- German J. Bloom's syndrome VIII Review of clinical and genetic aspects. In: Goodman RM, Motulsky AG, editors. Genetic diseases among Askenazi jews. New York: Raven Press; 1979. pp. 121–39.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
