

Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy
- PMID: 20179356
- PMCID: PMC2827951
- DOI: 10.1172/JCI40076
Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy
Erratum in
- J Clin Invest. 2010 Apr;120(4):1362. Jackson, Peter [corrected to Jackson, Peter K]
Abstract
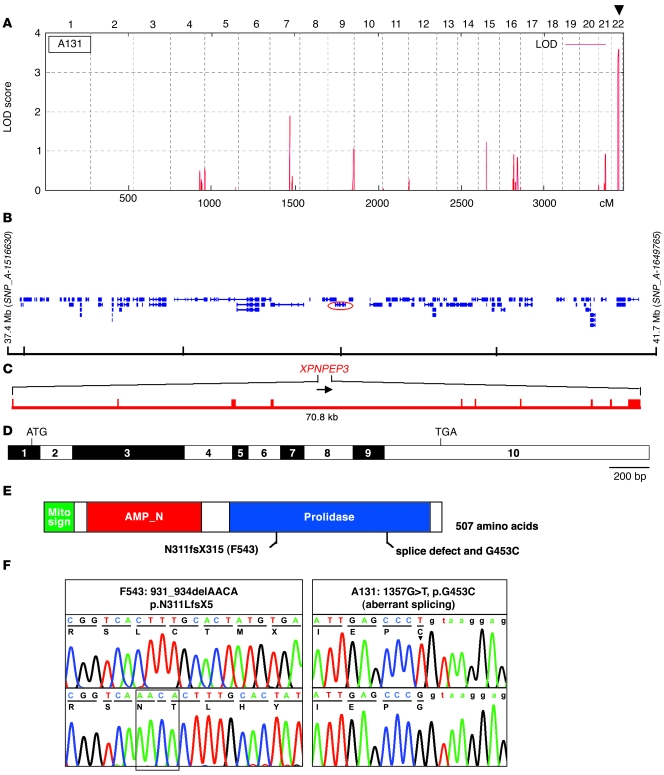
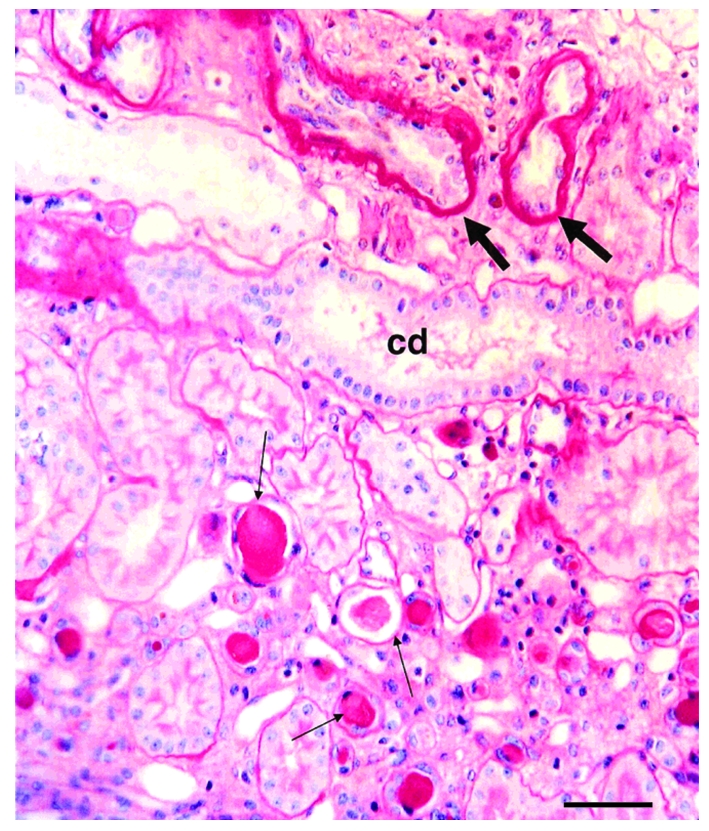
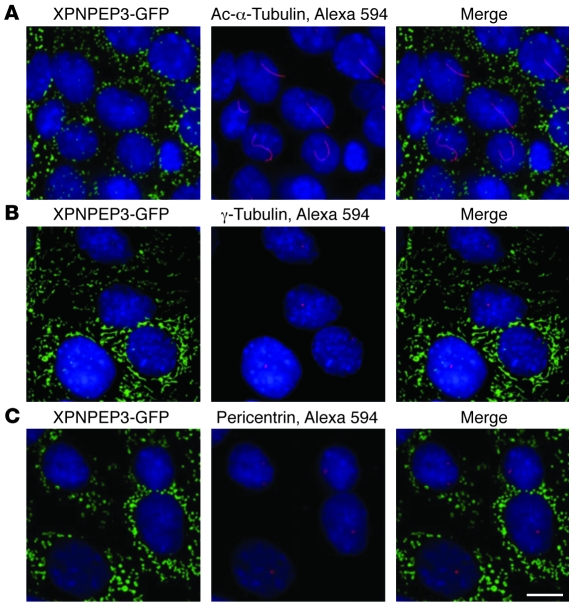
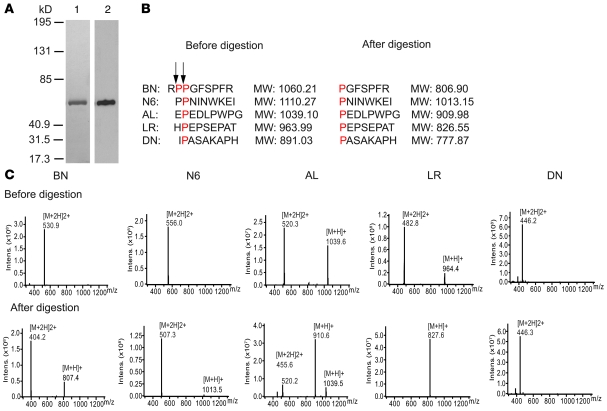
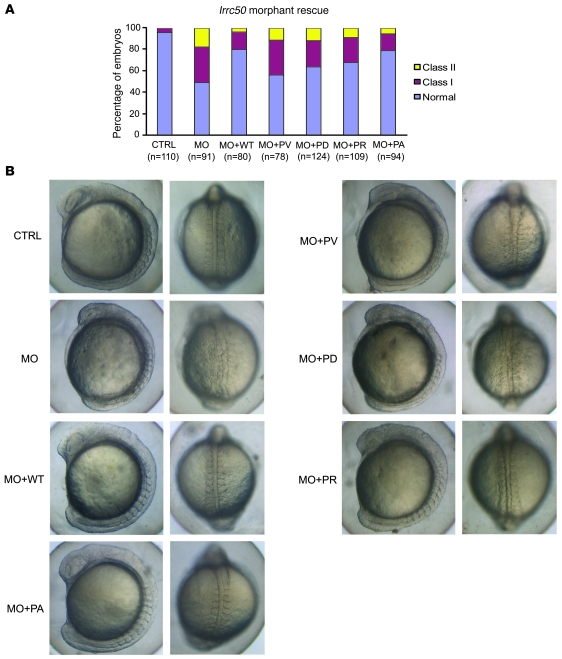
The autosomal recessive kidney disease nephronophthisis (NPHP) constitutes the most frequent genetic cause of terminal renal failure in the first 3 decades of life. Ten causative genes (NPHP1-NPHP9 and NPHP11), whose products localize to the primary cilia-centrosome complex, support the unifying concept that cystic kidney diseases are "ciliopathies". Using genome-wide homozygosity mapping, we report here what we believe to be a new locus (NPHP-like 1 [NPHPL1]) for an NPHP-like nephropathy. In 2 families with an NPHP-like phenotype, we detected homozygous frameshift and splice-site mutations, respectively, in the X-prolyl aminopeptidase 3 (XPNPEP3) gene. In contrast to all known NPHP proteins, XPNPEP3 localizes to mitochondria of renal cells. However, in vivo analyses also revealed a likely cilia-related function; suppression of zebrafish xpnpep3 phenocopied the developmental phenotypes of ciliopathy morphants, and this effect was rescued by human XPNPEP3 that was devoid of a mitochondrial localization signal. Consistent with a role for XPNPEP3 in ciliary function, several ciliary cystogenic proteins were found to be XPNPEP3 substrates, for which resistance to N-terminal proline cleavage resulted in attenuated protein function in vivo in zebrafish. Our data highlight an emerging link between mitochondria and ciliary dysfunction, and suggest that further understanding the enzymatic activity and substrates of XPNPEP3 will illuminate novel cystogenic pathways.
Figures

Comment in
-
Lights on for aminopeptidases in cystic kidney disease.J Clin Invest. 2010 Mar;120(3):660-3. doi: 10.1172/JCI42378. Epub 2010 Feb 22. J Clin Invest. 2010. PMID: 20179346 Free PMC article.
Similar articles
-
Lights on for aminopeptidases in cystic kidney disease.J Clin Invest. 2010 Mar;120(3):660-3. doi: 10.1172/JCI42378. Epub 2010 Feb 22. J Clin Invest. 2010. PMID: 20179346 Free PMC article.
-
Structure of the human aminopeptidase XPNPEP3 and comparison of its in vitro activity with Icp55 orthologs: Insights into diverse cellular processes.J Biol Chem. 2017 Jun 16;292(24):10035-10047. doi: 10.1074/jbc.M117.783357. Epub 2017 May 5. J Biol Chem. 2017. PMID: 28476889 Free PMC article.
-
Whole Exome Sequencing Reveals a XPNPEP3 Novel Mutation Causing Nephronophthisis in a Pediatric Patient.Iran Biomed J. 2020 Nov;24(6):405-8. doi: 10.29252/ibj.24.6.400. Epub 2020 May 31. Iran Biomed J. 2020. PMID: 32660933 Free PMC article.
-
Novel mutation in XPNPEP3 in a patient with heart failure without nephronophthisis-like nephropathy (NPHPL1): case report and literature review.BMC Pediatr. 2024 Oct 4;24(1):632. doi: 10.1186/s12887-024-05124-z. BMC Pediatr. 2024. PMID: 39363162 Free PMC article. Review.
-
Nephronophthisis-associated ciliopathies.J Am Soc Nephrol. 2007 Jun;18(6):1855-71. doi: 10.1681/ASN.2006121344. Epub 2007 May 18. J Am Soc Nephrol. 2007. PMID: 17513324 Review.
Cited by
-
Focus on Mitochondrial Respiratory Chain: Potential Therapeutic Target for Chronic Renal Failure.Int J Mol Sci. 2024 Jan 12;25(2):949. doi: 10.3390/ijms25020949. Int J Mol Sci. 2024. PMID: 38256023 Free PMC article. Review.
-
The ciliopathies: a transitional model into systems biology of human genetic disease.Curr Opin Genet Dev. 2012 Jun;22(3):290-303. doi: 10.1016/j.gde.2012.04.006. Epub 2012 May 23. Curr Opin Genet Dev. 2012. PMID: 22632799 Free PMC article. Review.
-
The ciliary transition zone: from morphology and molecules to medicine.Trends Cell Biol. 2012 Apr;22(4):201-10. doi: 10.1016/j.tcb.2012.02.001. Epub 2012 Mar 6. Trends Cell Biol. 2012. PMID: 22401885 Free PMC article. Review.
-
Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy.J Med Genet. 2011 Feb;48(2):105-16. doi: 10.1136/jmg.2010.082552. Epub 2010 Nov 10. J Med Genet. 2011. PMID: 21068128 Free PMC article.
-
The transcriptional response of skin to fluorescent light exposure in viviparous (Xiphophorus) and oviparous (Danio, Oryzias) fishes.Comp Biochem Physiol C Toxicol Pharmacol. 2018 Jun;208:77-86. doi: 10.1016/j.cbpc.2017.10.003. Epub 2017 Oct 7. Comp Biochem Physiol C Toxicol Pharmacol. 2018. PMID: 29017858 Free PMC article.
References
-
- Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18(6):1855–1871. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DK1068306/DK/NIDDK NIH HHS/United States
- EY007961/EY/NEI NIH HHS/United States
- R01 DK072301/DK/NIDDK NIH HHS/United States
- MOP-378061/CAPMC/ CIHR/Canada
- R01 DK068306/DK/NIDDK NIH HHS/United States
- R01 EY007961/EY/NEI NIH HHS/United States
- R01 DK075972/DK/NIDDK NIH HHS/United States
- K08 DK071108/DK/NIDDK NIH HHS/United States
- K12 HD000850/HD/NICHD NIH HHS/United States
- DK079541/DK/NIDDK NIH HHS/United States
- F32 DK079541/DK/NIDDK NIH HHS/United States
- DK064614/DK/NIDDK NIH HHS/United States
- R01 HD042601/HD/NICHD NIH HHS/United States
- R01 DK064614/DK/NIDDK NIH HHS/United States
- R01DK075972/DK/NIDDK NIH HHS/United States
- DK1069274/DK/NIDDK NIH HHS/United States
- R01HD04260/HD/NICHD NIH HHS/United States
- R01DK072301/DK/NIDDK NIH HHS/United States
- DK071108/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
