

Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome)
- PMID: 19224584
- PMCID: PMC3048808
- DOI: 10.1002/humu.20828
Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome)
Abstract
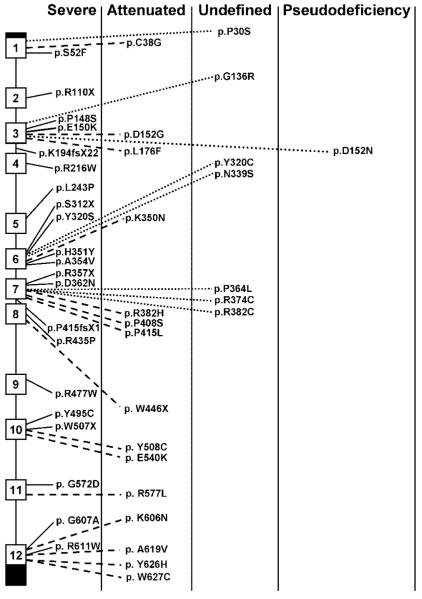
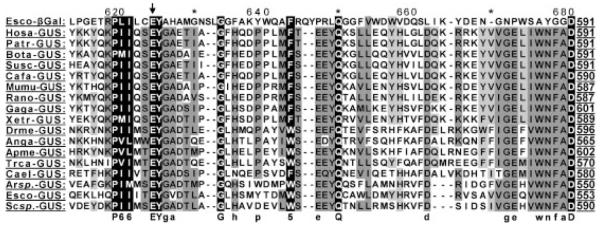
Mucopolysaccharidosis VII (MPS VII; Sly syndrome) is an autosomal recessive disorder caused by a deficiency of beta-glucuronidase (GUS, EC 3.2.1.31; GUSB). GUS is required to degrade glycosaminoglycans (GAGs), including heparan sulfate (HS), dermatan sulfate (DS), and chondroitin-4,6-sulfate (CS). Accumulation of undegraded GAGs in lysosomes of affected tissues leads to mental retardation, short stature, hepatosplenomegaly, bone dysplasia, and hydrops fetalis. We summarize information on the 49 unique, disease-causing mutations determined so far in the GUS gene, including nine novel mutations (eight missense and one splice-site). This heterogeneity in GUS gene mutations contributes to the extensive clinical variability among patients with MPS VII. One pseudodeficiency allele, one polymorphism causing an amino acid change, and one silent variant in the coding region are also described. Among the 103 analyzed mutant alleles, missense mutations accounted for 78.6%; nonsense mutations, 12.6%; deletions, 5.8%; and splice-site mutations, 2.9%. Transitional mutations at CpG dinucleotides made up 40.8% of all the described mutations. The five most frequent mutations (accounting for 44/103 alleles) were exonic point mutations, p.L176F, p.R357X, p.P408S, p.P415L, and p.A619 V. Genotype/phenotype correlation was attempted by correlating the effects of certain missense mutations or enzyme activity and stability within phenotypes. These were in turn correlated with the location of the mutation in the tertiary structure of GUS. A total of seven murine, one feline, and one canine model of MPS VII have been characterized for phenotype and genotype.
(c) 2009 Wiley-Liss, Inc.
Figures
Similar articles
-
Missense models [Gustm(E536A)Sly, Gustm(E536Q)Sly, and Gustm(L175F)Sly] of murine mucopolysaccharidosis type VII produced by targeted mutagenesis.Proc Natl Acad Sci U S A. 2002 Nov 12;99(23):14982-7. doi: 10.1073/pnas.232570999. Epub 2002 Oct 28. Proc Natl Acad Sci U S A. 2002. PMID: 12403825 Free PMC article.
-
Molecular analysis of patients with beta-glucuronidase deficiency presenting as hydrops fetalis or as early mucopolysaccharidosis VII.Am J Hum Genet. 1996 Mar;58(3):457-71. Am J Hum Genet. 1996. PMID: 8644704 Free PMC article.
-
Lentiviral-mediated gene therapy results in sustained expression of β-glucuronidase for up to 12 months in the gus(mps/mps) and up to 18 months in the gus(tm(L175F)Sly) mouse models of mucopolysaccharidosis type VII.Hum Gene Ther. 2014 Sep;25(9):798-810. doi: 10.1089/hum.2013.141. Epub 2014 Aug 14. Hum Gene Ther. 2014. PMID: 25003807
-
Mucopolysaccharidosis type VII (Sly syndrome) - What do we know?Mol Genet Metab. 2024 Mar;141(3):108145. doi: 10.1016/j.ymgme.2024.108145. Epub 2024 Jan 17. Mol Genet Metab. 2024. PMID: 38301529 Review.
-
Murine mucopolysaccharidosis VIL: impact of therapies on the phenotype, clinical course, and pathology in a model of a lysosomal storage disease.Pediatr Dev Pathol. 2001 Sep-Oct;4(5):421-33. doi: 10.1007/s10024001-0079-1. Pediatr Dev Pathol. 2001. PMID: 11779044 Review.
Cited by
-
Diagnosis and Emerging Treatment Strategies for Mucopolysaccharidosis VII (Sly Syndrome).Ther Clin Risk Manag. 2022 Dec 22;18:1143-1155. doi: 10.2147/TCRM.S351300. eCollection 2022. Ther Clin Risk Manag. 2022. PMID: 36578769 Free PMC article. Review.
-
First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation.Int J Mol Sci. 2019 Oct 28;20(21):5345. doi: 10.3390/ijms20215345. Int J Mol Sci. 2019. PMID: 31661765 Free PMC article.
-
Polymer-based drug delivery systems under investigation for enzyme replacement and other therapies of lysosomal storage disorders.Adv Drug Deliv Rev. 2023 Jun;197:114683. doi: 10.1016/j.addr.2022.114683. Epub 2023 Jan 16. Adv Drug Deliv Rev. 2023. PMID: 36657645 Free PMC article. Review.
-
Non-progressive Nonimmune Hydrops Fetalis Caused by a Novel Mutation in GUSB Gene.Iran J Child Neurol. 2020 Spring;14(2):101-106. Iran J Child Neurol. 2020. PMID: 32256629 Free PMC article.
-
Corneal Regeneration Using Gene Therapy Approaches.Cells. 2023 Apr 28;12(9):1280. doi: 10.3390/cells12091280. Cells. 2023. PMID: 37174680 Free PMC article. Review.
References
-
- Antonarakis SE, Krawczak M, Cooper DN. The nature and mechanisms of human gene mutation. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. pp. 343–377.
-
- Brot FE, Bell CE, Jr, Sly WS. Purification and properties of beta-glucuronidase from human placenta. Biochemistry. 1978;17:385–391. - PubMed
-
- Fukuda S, Tomatsu S, Sukegawa K, Sasaki T, Yamada Y, Kuwahara T, Okamoto H, Ikedo Y, Yamaguchi S, Orii T. Molecular analysis of mucopolysaccharidosis type VII. J Inherit Metab Dis. 1991;14:800–804. - PubMed
-
- Fyfe JC, Kurzhals RL, Lassaline ME, Henthorn PS, Alur PR, Wang P, Wolfe JH, Giger U, Haskins ME, Patterson DF, Sun H, Jain S, Yuhki N. Molecular basis of feline beta-glucuronidase deficiency: an animal model of mucopolysaccharidosis VII. Genomics. 1999;58:121–128. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
