

Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4
- PMID: 17999358
- PMCID: PMC2276340
- DOI: 10.1086/522090
Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4
Abstract
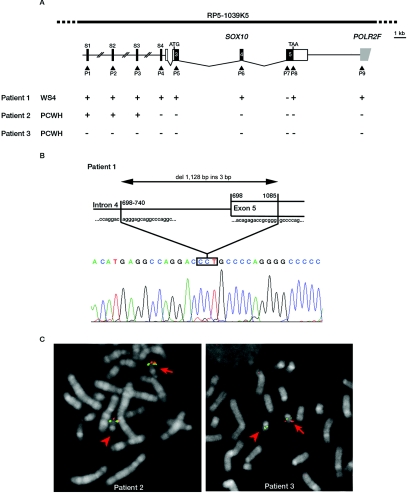
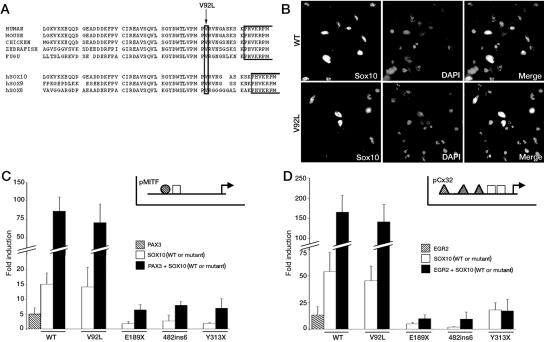
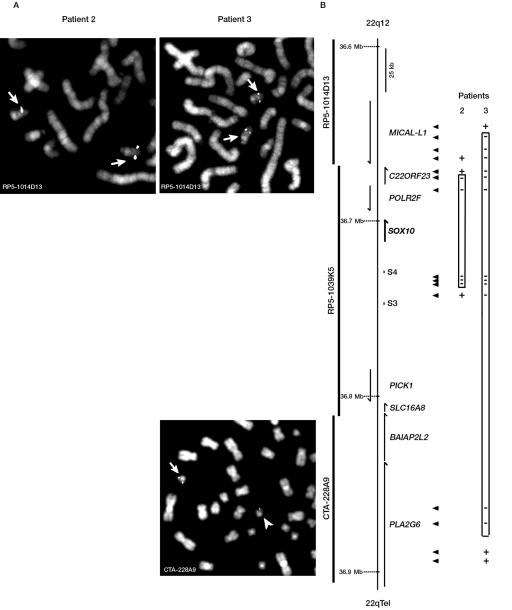
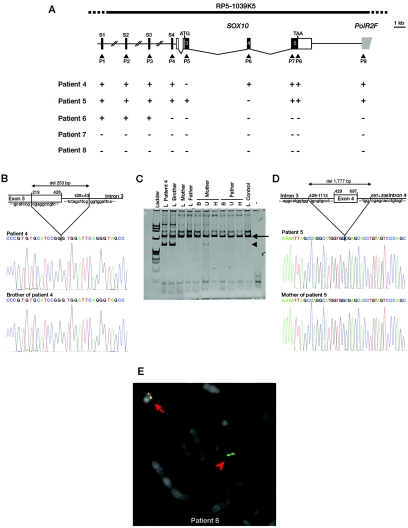
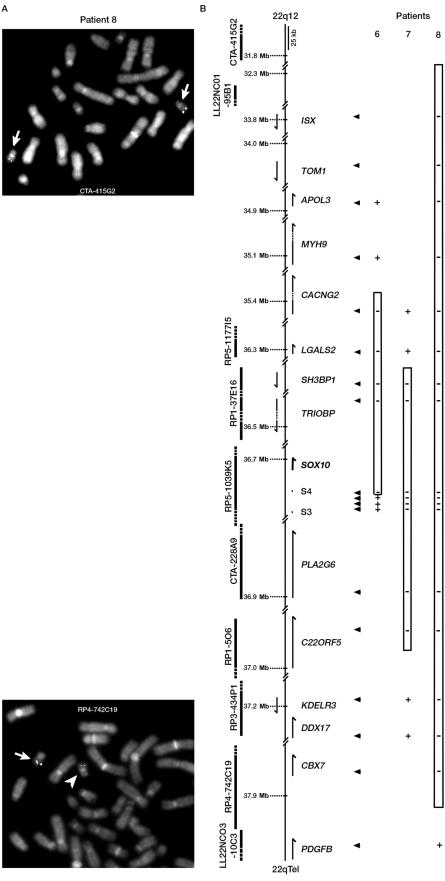
Waardenburg syndrome (WS) is an auditory-pigmentary disorder that exhibits varying combinations of sensorineural hearing loss and abnormal pigmentation of the hair and skin. Depending on additional symptoms, WS is classified into four subtypes, WS1-WS4. Absence of additional features characterizes WS2. The association of facial dysmorphic features defines WS1 and WS3, whereas the association with Hirschsprung disease (aganglionic megacolon) characterizes WS4, also called "Waardenburg-Hirschsprung disease." Mutations within the genes MITF and SNAI2 have been identified in WS2, whereas mutations of EDN3, EDNRB, and SOX10 have been observed in patients with WS4. However, not all cases are explained at the molecular level, which raises the possibility that other genes are involved or that some mutations within the known genes are not detected by commonly used genotyping methods. We used a combination of semiquantitative fluorescent multiplex polymerase chain reaction and fluorescent in situ hybridization to search for SOX10 heterozygous deletions. We describe the first characterization of SOX10 deletions in patients presenting with WS4. We also found SOX10 deletions in WS2 cases, making SOX10 a new gene of WS2. Interestingly, neurological phenotypes reminiscent of that observed in WS4 (PCWH syndrome [peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, WS, and Hirschsprung disease]) were observed in some WS2-affected patients with SOX10 deletions. This study further characterizes the molecular complexity and the close relationship that links the different subtypes of WS.
Figures
Similar articles
-
Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome.Hum Mol Genet. 2000 Aug 12;9(13):1907-17. doi: 10.1093/hmg/9.13.1907. Hum Mol Genet. 2000. PMID: 10942418
-
Waardenburg syndrome type 4: report of two new cases caused by SOX10 mutations in Spain.Am J Med Genet A. 2014 Feb;164A(2):542-7. doi: 10.1002/ajmg.a.36302. Epub 2013 Dec 5. Am J Med Genet A. 2014. PMID: 24311220
-
Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature.Eur J Paediatr Neurol. 2006 Jan;10(1):11-7. doi: 10.1016/j.ejpn.2005.10.004. Epub 2006 Feb 28. Eur J Paediatr Neurol. 2006. PMID: 16504559
-
Mouse models for four types of Waardenburg syndrome.Pigment Cell Res. 2003 Oct;16(5):448-54. doi: 10.1034/j.1600-0749.2003.00066.x. Pigment Cell Res. 2003. PMID: 12950719 Review.
-
[Hereditary hypomelanocytoses: the role of PAX3, SOX10, MITF, SNAI2, KIT, EDN3 and EDNRB genes].Postepy Hig Med Dosw (Online). 2013 Nov 26;67:1109-18. doi: 10.5604/17322693.1077722. Postepy Hig Med Dosw (Online). 2013. PMID: 24379252 Review. Polish.
Cited by
-
Primary anorectal melanoma: an update.J Cancer. 2012;3:449-53. doi: 10.7150/jca.5187. Epub 2012 Oct 20. J Cancer. 2012. PMID: 23193431 Free PMC article.
-
Waardenburg Syndrome: The Contribution of Next-Generation Sequencing to the Identification of Novel Causative Variants.Audiol Res. 2023 Dec 21;14(1):9-25. doi: 10.3390/audiolres14010002. Audiol Res. 2023. PMID: 38391765 Free PMC article.
-
Spectrum of temporal bone abnormalities in patients with Waardenburg syndrome and SOX10 mutations.AJNR Am J Neuroradiol. 2013 Jun-Jul;34(6):1257-63. doi: 10.3174/ajnr.A3367. Epub 2012 Dec 13. AJNR Am J Neuroradiol. 2013. PMID: 23237859 Free PMC article.
-
A rapid improved multiplex ligation detection reaction method for the identification of gene mutations in hereditary hearing loss.PLoS One. 2019 Apr 11;14(4):e0215212. doi: 10.1371/journal.pone.0215212. eCollection 2019. PLoS One. 2019. PMID: 30973918 Free PMC article.
-
Deafness-in-a-dish: modeling hereditary deafness with inner ear organoids.Hum Genet. 2022 Apr;141(3-4):347-362. doi: 10.1007/s00439-021-02325-9. Epub 2021 Aug 3. Hum Genet. 2022. PMID: 34342719 Free PMC article. Review.
References
Web Resources
-
- BACPAC Resources, http://bacpac.chori.org/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for accession numbers NC_000021.7, NC_000023.9, and NC_000022.9)
-
- HMMgene, http://www.cbs.dtu.dk/services/HMMgene/ (for gene-structure prediction)
-
- Netgene2, http://www.cbs.dtu.dk/services/NetGene2/ (for neural-network predictions of splice sites in human DNA)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for WS1–WS4) - PubMed
References
-
- Le Douarin NM, Kalcheim C (1999) The neural crest. Cambridge University Press, Cambridge, United Kingdom
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
