

Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders
- PMID: 17332895
- PMCID: PMC1804361
- DOI: 10.1172/JCI29089
Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders
Abstract
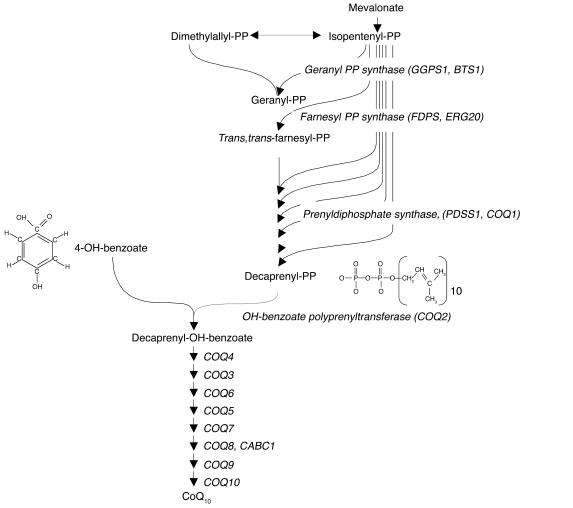
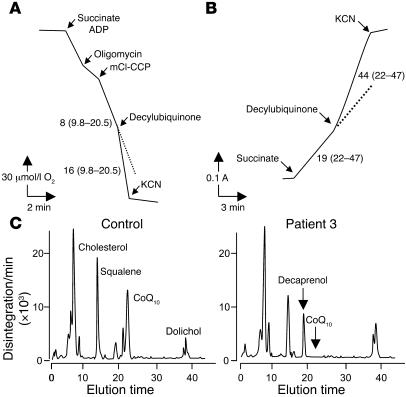
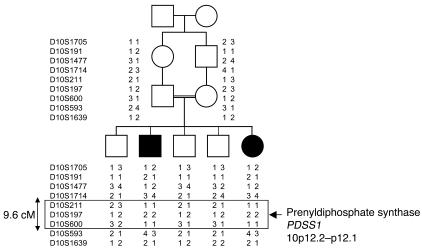
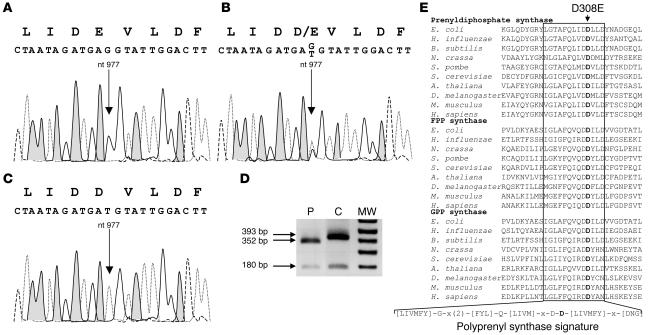
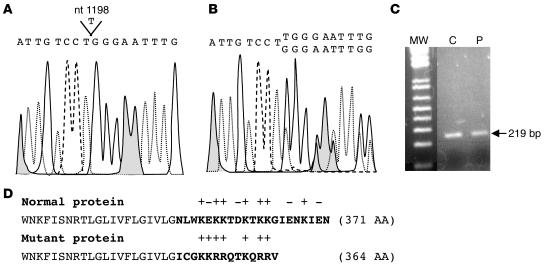
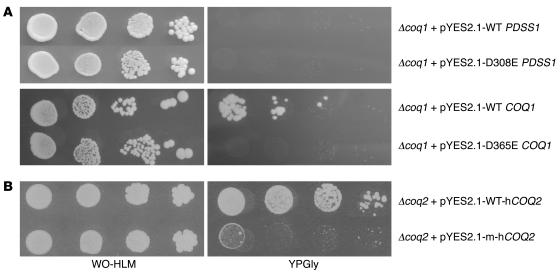
Coenzyme Q10 (CoQ10) plays a pivotal role in oxidative phosphorylation (OXPHOS), as it distributes electrons among the various dehydrogenases and the cytochrome segments of the respiratory chain. We have identified 2 novel inborn errors of CoQ10 biosynthesis in 2 distinct families. In both cases, enzymologic studies showed that quinone-dependent OXPHOS activities were in the range of the lowest control values, while OXPHOS enzyme activities were normal. CoQ10 deficiency was confirmed by restoration of normal OXPHOS activities after addition of quinone. A genome-wide search for homozygosity in family 1 identified a region of chromosome 10 encompassing the gene prenyldiphosphate synthase, subunit 1 (PDSS1), which encodes the human ortholog of the yeast COQ1 gene, a key enzyme of CoQ10 synthesis. Sequencing of PDSS1 identified a homozygous nucleotide substitution modifying a conserved amino acid of the protein (D308E). In the second family, direct sequencing of OH-benzoate polyprenyltransferase (COQ2), the human ortholog of the yeast COQ2 gene, identified a single base pair frameshift deletion resulting in a premature stop codon (c.1198delT, N401fsX415). Transformation of yeast Deltacoq1 and Deltacoq2 strains by mutant yeast COQ1 and mutant human COQ2 genes, respectively, resulted in defective growth on respiratory medium, indicating that these mutations are indeed the cause of OXPHOS deficiency.
Figures
Comment in
-
Mutations in coenzyme Q10 biosynthetic genes.J Clin Invest. 2007 Mar;117(3):587-9. doi: 10.1172/JCI31423. J Clin Invest. 2007. PMID: 17332886 Free PMC article.
Similar articles
-
Infantile and pediatric quinone deficiency diseases.Mitochondrion. 2007 Jun;7 Suppl:S112-21. doi: 10.1016/j.mito.2007.02.008. Epub 2007 Mar 16. Mitochondrion. 2007. PMID: 17442627 Review.
-
Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2).Eur J Paediatr Neurol. 2013 Nov;17(6):625-30. doi: 10.1016/j.ejpn.2013.05.013. Epub 2013 Jun 28. Eur J Paediatr Neurol. 2013. PMID: 23816342
-
Functional characterization of OsPPT1, which encodes p-hydroxybenzoate polyprenyltransferase involved in ubiquinone biosynthesis in Oryza sativa.Plant Cell Physiol. 2006 May;47(5):581-90. doi: 10.1093/pcp/pcj025. Epub 2006 Feb 24. Plant Cell Physiol. 2006. PMID: 16501255
-
COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement.J Am Soc Nephrol. 2007 Oct;18(10):2773-80. doi: 10.1681/ASN.2006080833. Epub 2007 Sep 12. J Am Soc Nephrol. 2007. PMID: 17855635
-
A novel mutation in COQ2 leading to fatal infantile multisystem disease.J Neurol Sci. 2013 Mar 15;326(1-2):24-8. doi: 10.1016/j.jns.2013.01.004. Epub 2013 Jan 21. J Neurol Sci. 2013. PMID: 23343605 Review.
Cited by
-
Update on clinical aspects and treatment of selected vitamin-responsive disorders II (riboflavin and CoQ 10).J Inherit Metab Dis. 2012 Jul;35(4):679-87. doi: 10.1007/s10545-011-9434-1. Epub 2012 Jan 10. J Inherit Metab Dis. 2012. PMID: 22231380 Review.
-
Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels.J Cell Biol. 2015 Feb 16;208(4):429-42. doi: 10.1083/jcb.201411100. J Cell Biol. 2015. PMID: 25688136 Free PMC article.
-
An update on the role of ferroptosis in the pathogenesis of osteoporosis.EFORT Open Rev. 2024 Aug 1;9(8):712-722. doi: 10.1530/EOR-23-0148. EFORT Open Rev. 2024. PMID: 39087516 Free PMC article. Review.
-
A hypoxia-related signature for clinically predicting diagnosis, prognosis and immune microenvironment of hepatocellular carcinoma patients.J Transl Med. 2020 Sep 4;18(1):342. doi: 10.1186/s12967-020-02492-9. J Transl Med. 2020. PMID: 32887635 Free PMC article.
-
COQ6 mutation in patients with nephrotic syndrome, sensorineural deafness, and optic atrophy.JIMD Rep. 2020 May 5;54(1):37-44. doi: 10.1002/jmd2.12068. eCollection 2020 Jul. JIMD Rep. 2020. PMID: 32685349 Free PMC article.
References
-
- Turunen M., Olsson J., Dallner G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta. 2004;1660:171–199. - PubMed
-
- Sobreira C., et al. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238–1243. - PubMed
-
- Boitier E., et al. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J. Neurol. Sci. 1998;156:41–46. - PubMed
-
- Di Giovanni S., et al. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology. 2001;57:515–518. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
