

Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity
- PMID: 16602101
- PMCID: PMC2652706
- DOI: 10.1002/ajmg.c.30089
Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity
Abstract
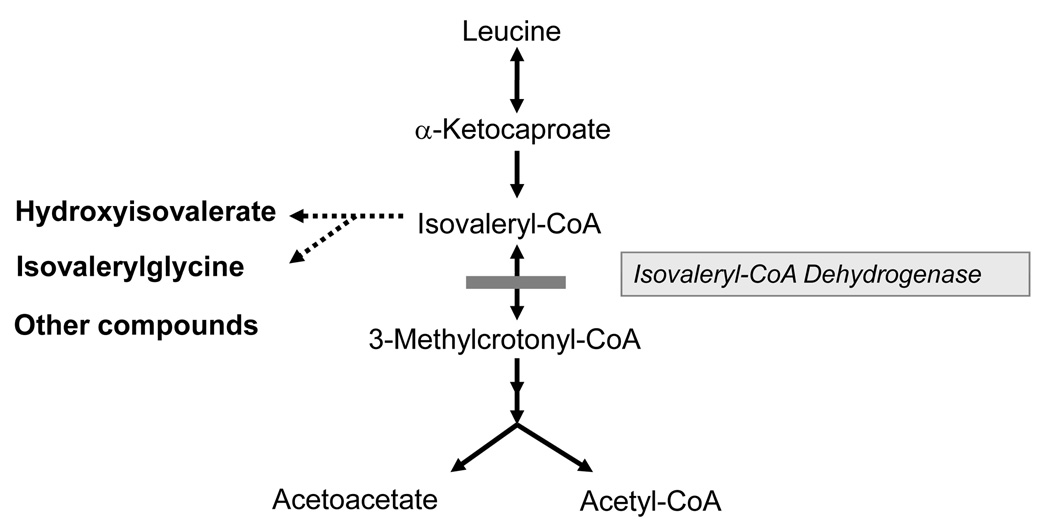
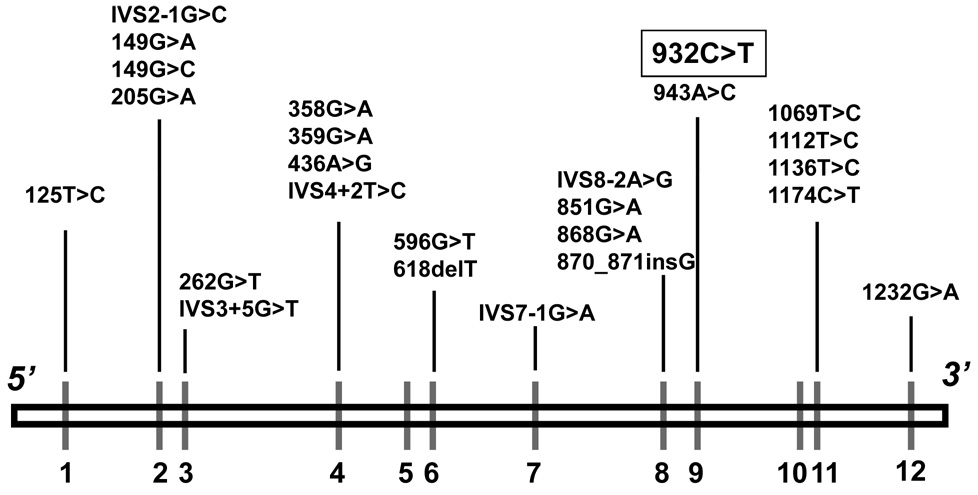
Isovaleric acidemia (IVA) is an autosomal recessive inborn error of leucine metabolism caused by a deficiency of the mitochondrial enzyme isovaleryl-CoA dehydrogenase (IVD) resulting in the accumulation of derivatives of isovaleryl-CoA. It was the first organic acidemia recognized in humans and can cause significant morbidity and mortality. Early diagnosis and treatment with a protein restricted diet and supplementation with carnitine and glycine are effective in promoting normal development in severely affected individuals. Both intra- and interfamilial variability have been recognized. Initially, two phenotypes with either an acute neonatal or a chronic intermittent presentation were described. More recently, a third group of individuals with mild biochemical abnormalities who can be asymptomatic have been identified through newborn screening of blood spots by tandem mass spectrometry. IVD is a flavoenzyme that catalyzes the conversion of isovaleryl-CoA to 3-methylcrotonyl-CoA and transfers electrons to the electron transfer flavoprotein. Human IVD has been purified from tissue and recombinant sources and its biochemical and physical properties have been extensively studied. Molecular analysis of the IVD gene from patients with IVA has allowed characterization of different types of mutations in this gene. One missense mutation, 932C>T (A282V), is particularly common in patients identified through newborn screening with mild metabolite elevations and who have remained asymptomatic to date. This mutation leads to a partially active enzyme with altered catalytic properties; however, its effects on clinical outcome and the necessity of therapy are still unknown. A better understanding of the heterogeneity of this disease and the relevance of genotype/phenotype correlations to clinical management of patients are among the challenges remaining in the study of this disorder in the coming years.
(c) 2006 Wiley-Liss, Inc.
Figures
Similar articles
-
Aspects of Newborn Screening in Isovaleric Acidemia.Int J Neonatal Screen. 2018 Jan 29;4(1):7. doi: 10.3390/ijns4010007. eCollection 2018 Mar. Int J Neonatal Screen. 2018. PMID: 33072933 Free PMC article. Review.
-
Phenotypic Variability and Newly Identified Mutations of the IVD Gene in Japanese Patients with Isovaleric Acidemia.Tohoku J Exp Med. 2015 Jun;236(2):103-6. doi: 10.1620/tjem.236.103. Tohoku J Exp Med. 2015. PMID: 26018748
-
A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening.Am J Hum Genet. 2004 Dec;75(6):1136-42. doi: 10.1086/426318. Epub 2004 Oct 14. Am J Hum Genet. 2004. PMID: 15486829 Free PMC article.
-
Characterization of variants of uncertain significance in isovaleryl-CoA dehydrogenase identified through newborn screening: An approach for faster analysis.Mol Genet Metab. 2021 Sep-Oct;134(1-2):29-36. doi: 10.1016/j.ymgme.2021.08.012. Epub 2021 Aug 30. Mol Genet Metab. 2021. PMID: 34535384 Free PMC article.
-
Molecular basis of isovaleric acidemia and medium-chain acyl-CoA dehydrogenase deficiency.Enzyme. 1987;38(1-4):91-107. doi: 10.1159/000469195. Enzyme. 1987. PMID: 3326738 Review.
Cited by
-
Aspects of Newborn Screening in Isovaleric Acidemia.Int J Neonatal Screen. 2018 Jan 29;4(1):7. doi: 10.3390/ijns4010007. eCollection 2018 Mar. Int J Neonatal Screen. 2018. PMID: 33072933 Free PMC article. Review.
-
Atypical MR lenticular signal change in infantile isovaleric acidemia.Indian J Radiol Imaging. 2016 Jan-Mar;26(1):131-4. doi: 10.4103/0971-3026.178362. Indian J Radiol Imaging. 2016. PMID: 27081237 Free PMC article.
-
Identifying potential dietary treatments for inherited metabolic disorders using Drosophila nutrigenomics.Cell Rep. 2024 Mar 26;43(3):113861. doi: 10.1016/j.celrep.2024.113861. Epub 2024 Feb 27. Cell Rep. 2024. PMID: 38416643 Free PMC article.
-
The glycine N-acyltransferases, GLYAT and GLYATL1, contribute to the detoxification of isovaleryl-CoA - an in-silico and in vitro validation.Comput Struct Biotechnol J. 2023 Jan 31;21:1236-1248. doi: 10.1016/j.csbj.2023.01.041. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 36817957 Free PMC article.
-
Deploying an In Vitro Gut Model to Assay the Impact of the Mannan-Oligosaccharide Prebiotic Bio-Mos on the Atlantic Salmon (Salmo salar) Gut Microbiome.Microbiol Spectr. 2022 Jun 29;10(3):e0195321. doi: 10.1128/spectrum.01953-21. Epub 2022 May 9. Microbiol Spectr. 2022. PMID: 35532227 Free PMC article.
References
-
- Abdenur JE, Chamoles NA, Guinle AE, Schenone AB, Fuertes AN. Diagnosis of isovaleric acidaemia by tandem mass spectrometry: false positive result due to pivaloylcarnitine in a newborn screening programme. J Inherit Metab Dis. 1998;21:624–630. - PubMed
-
- Andresen B, Christensen E, Corydon T, Bross P, Pilgaard B, Wanders R, Ruiter J, Simonsen H, Winter V, Knudsen I, Schroeder LD, Gregersen N, Skovby F. Isolated 2-methylbutyrylglycinuria caused short/branched-chain acyl-CoA dehydrogenase deficiency: Identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism. Am J Hum Gen. 2000;67:1095–1103. - PMC - PubMed
-
- Arnold WC, Brewster M, Byrne WJ, Booth B. Fanconi syndrome in a patient with a variant of isovaleric acidemia. Intl J Pediatr Nephr. 1986;7:95–98. - PubMed
-
- Attia N, Sakati N, al Ashwal A, al Saif R, Rashed M, Ozand PT. Isovaleric academia appearing as diabetic ketoacidosis. J Inherited Metab Disease. 1996;19:85–86. - PubMed
-
- Berry GT, Yudkoff M, Segal S. Isovaleric acidemia: medical and neurodevelopmental effects of long-term therapy. J Pediatr. 1988;113:58–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
