

Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia
- PMID: 14623864
- PMCID: PMC2173682
- DOI: 10.1083/jcb.200304112
Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia
Abstract
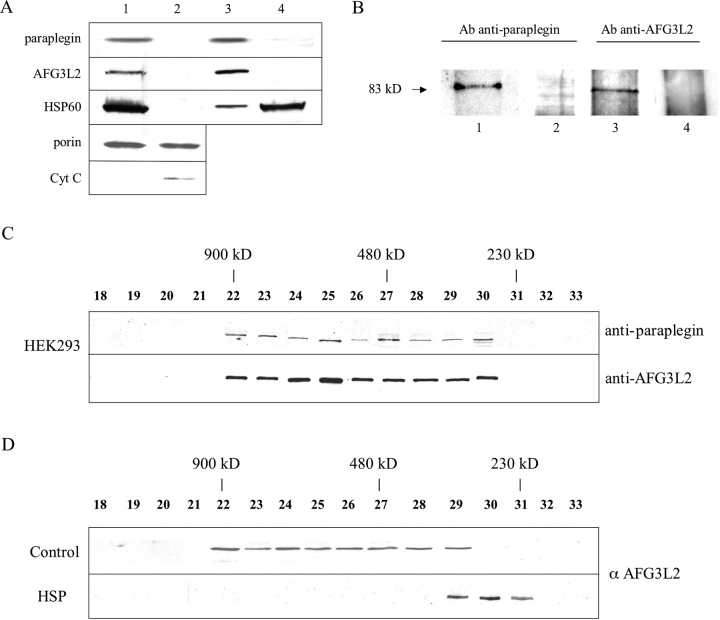
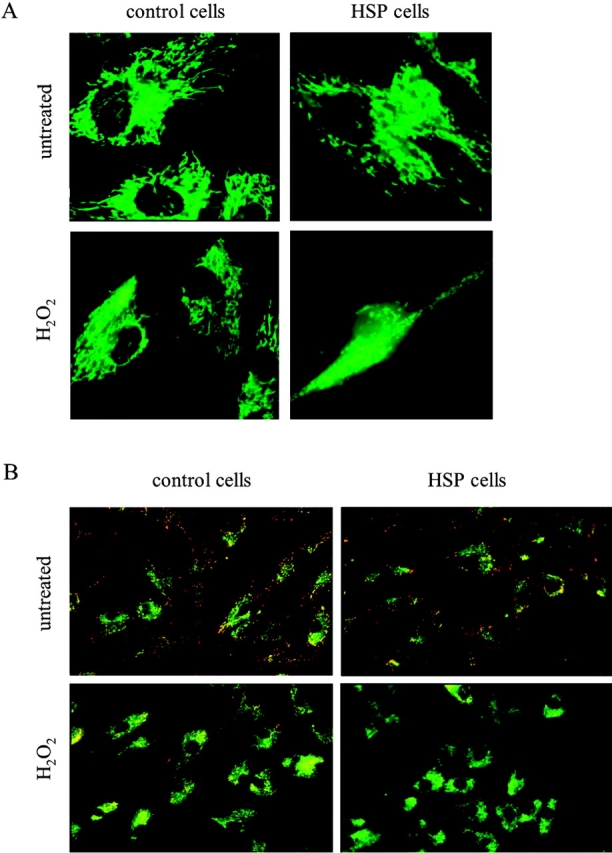
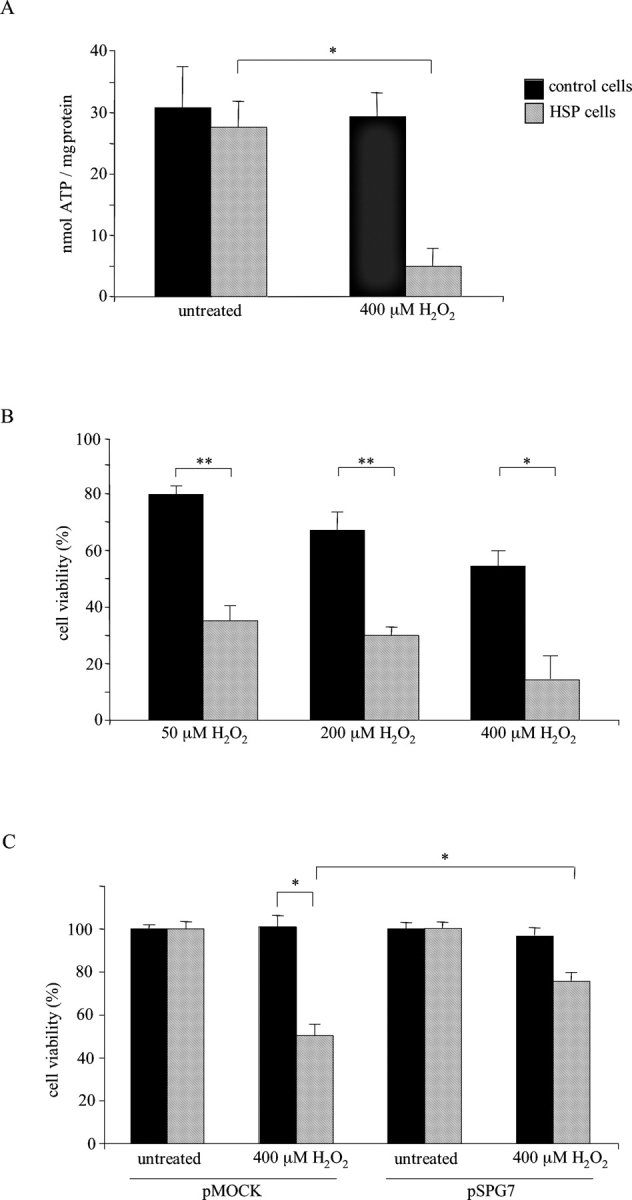
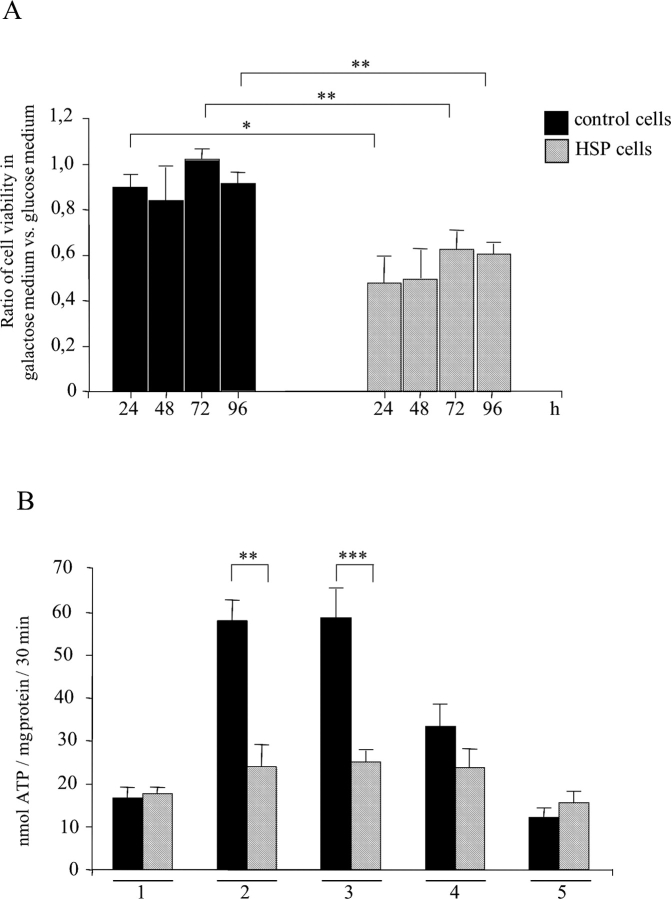
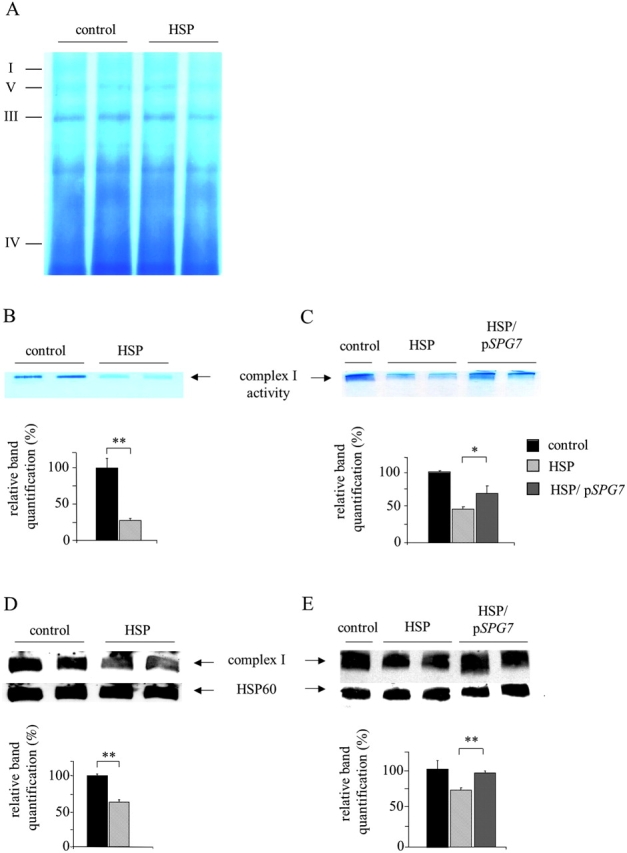
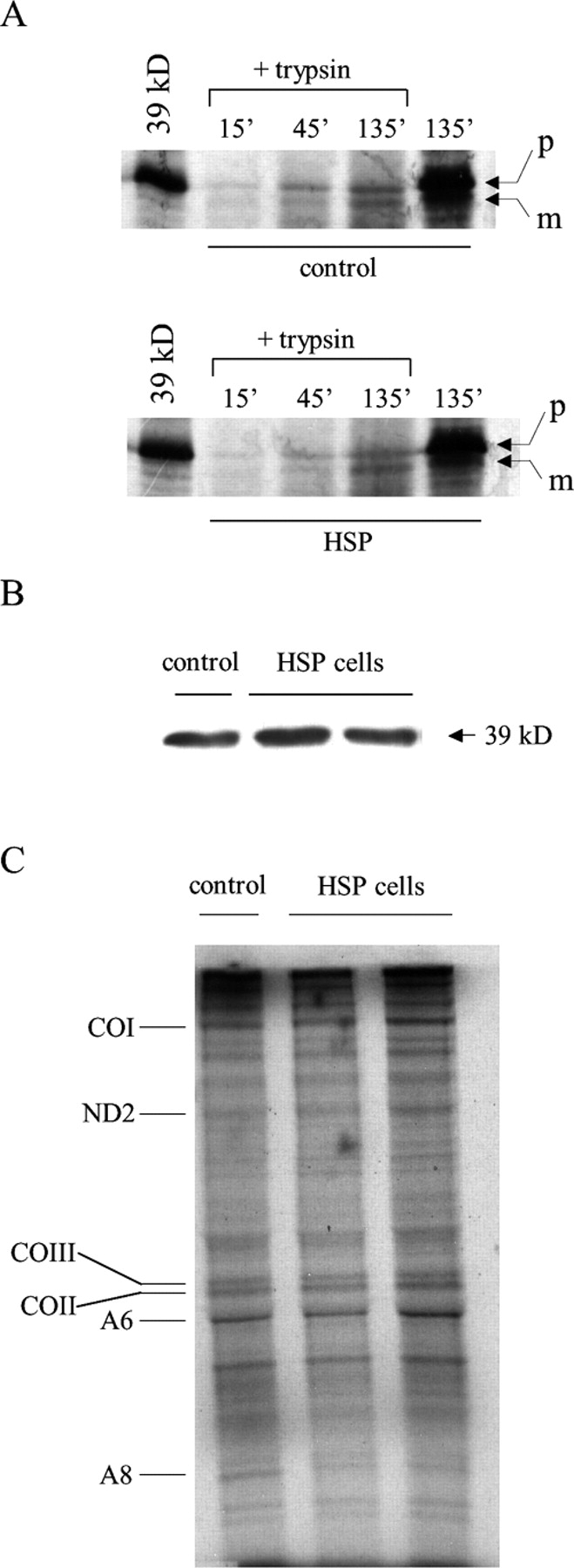
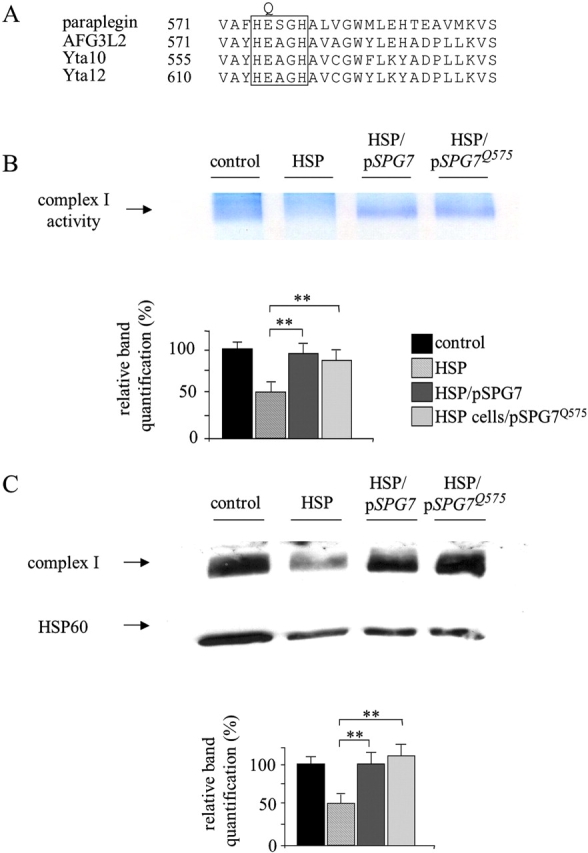
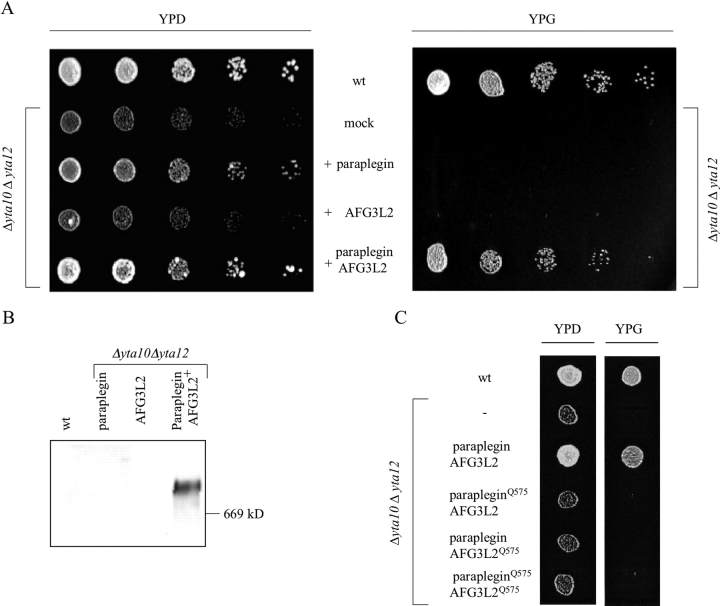
Mmutations in paraplegin, a putative mitochondrial metallopeptidase of the AAA family, cause an autosomal recessive form of hereditary spastic paraplegia (HSP). Here, we analyze the function of paraplegin at the cellular level and characterize the phenotypic defects of HSP patients' cells lacking this protein. We demonstrate that paraplegin coassembles with a homologous protein, AFG3L2, in the mitochondrial inner membrane. These two proteins form a high molecular mass complex, which we show to be aberrant in HSP fibroblasts. The loss of this complex causes a reduced complex I activity in mitochondria and an increased sensitivity to oxidant stress, which can both be rescued by exogenous expression of wild-type paraplegin. Furthermore, complementation studies in yeast demonstrate functional conservation of the human paraplegin-AFG3L2 complex with the yeast m-AAA protease and assign proteolytic activity to this structure. These results shed new light on the molecular pathogenesis of HSP and functionally link AFG3L2 to this neurodegenerative disease.
Figures
Similar articles
-
Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia.Mol Cell Biol. 2007 Jan;27(2):758-67. doi: 10.1128/MCB.01470-06. Epub 2006 Nov 13. Mol Cell Biol. 2007. PMID: 17101804 Free PMC article.
-
The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria.Cell. 2005 Oct 21;123(2):277-89. doi: 10.1016/j.cell.2005.08.003. Cell. 2005. PMID: 16239145
-
Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases.PLoS Genet. 2011 Oct;7(10):e1002325. doi: 10.1371/journal.pgen.1002325. Epub 2011 Oct 13. PLoS Genet. 2011. PMID: 22022284 Free PMC article.
-
Translating m-AAA protease function in mitochondria to hereditary spastic paraplegia.Trends Mol Med. 2006 Jun;12(6):262-9. doi: 10.1016/j.molmed.2006.04.002. Epub 2006 May 2. Trends Mol Med. 2006. PMID: 16647881 Review.
-
Hereditary spastic paraplegia: mitochondrial metalloproteases of yeast.Hum Genet. 1999 Jun;104(6):443-8. doi: 10.1007/s004390050985. Hum Genet. 1999. PMID: 10453730 Review.
Cited by
-
Hereditary spastic paraplegia.Curr Neurol Neurosci Rep. 2006 Jan;6(1):65-76. doi: 10.1007/s11910-996-0011-1. Curr Neurol Neurosci Rep. 2006. PMID: 16469273 Review.
-
OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria.Mol Biol Cell. 2007 Sep;18(9):3582-90. doi: 10.1091/mbc.e07-02-0164. Epub 2007 Jul 5. Mol Biol Cell. 2007. PMID: 17615298 Free PMC article.
-
Mouse brain expression patterns of Spg7, Afg3l1, and Afg3l2 transcripts, encoding for the mitochondrial m-AAA protease.BMC Neurosci. 2010 Apr 28;11:55. doi: 10.1186/1471-2202-11-55. BMC Neurosci. 2010. PMID: 20426821 Free PMC article.
-
Quality control of mitochondria: protection against neurodegeneration and ageing.EMBO J. 2008 Jan 23;27(2):306-14. doi: 10.1038/sj.emboj.7601972. EMBO J. 2008. PMID: 18216873 Free PMC article. Review.
-
A Novel SPG7 Gene Pathogenic Variant in a Cypriot Family With Autosomal Recessive Spastic Ataxia.Front Genet. 2022 Jan 13;12:812640. doi: 10.3389/fgene.2021.812640. eCollection 2021. Front Genet. 2022. PMID: 35096021 Free PMC article.
References
-
- Aguirre, T., L. Van Den Bosch, K. Goetschalckx, P. Tilkin, G. Mathijs, J.J. Cassiman, and W. Robberecht. 1998. Increased sensitivity of fibroblasts from amyotrophic lateral sclerosis patients to oxidative stress. Ann. Neurol. 43:452–457. - PubMed
-
- Arlt, H., R. Tauer, H. Feldmann, W. Neupert, and T. Langer. 1996. The YTA10-12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell. 85:875–885. - PubMed
-
- Banfi, S., M.T. Bassi, G. Andolfi, A. Marchitiello, S. Zanotta, A. Ballabio, G. Casari, and B. Franco. 1999. Identification and characterization of AFG3L2, a novel paraplegin-related gene. Genomics. 59:51–58. - PubMed
-
- Barrientos, A., and C.T. Moraes. 1999. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J. Biol. Chem. 274:16188–16197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
