

Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3
- PMID: 12621583
- PMCID: PMC1180354
- DOI: 10.1086/374320
Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3
Abstract
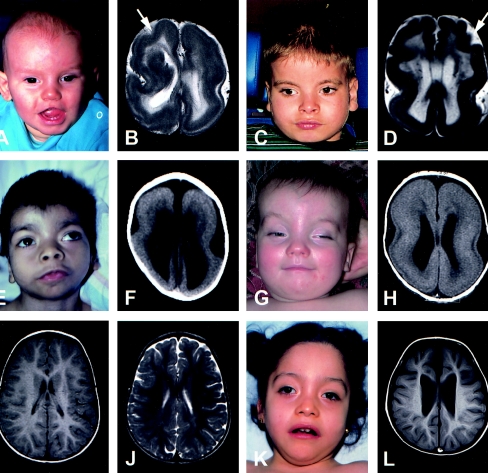
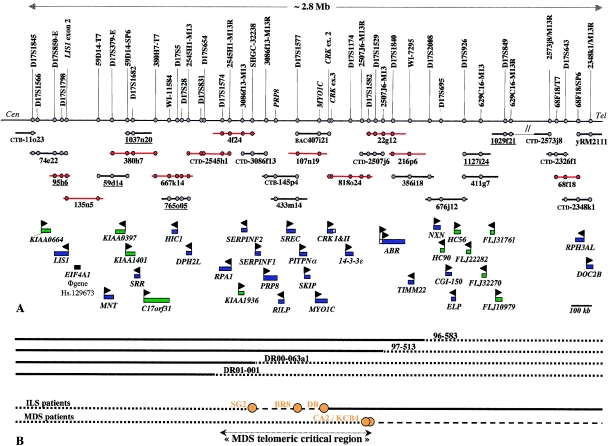
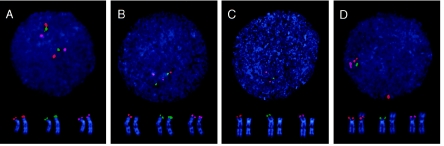
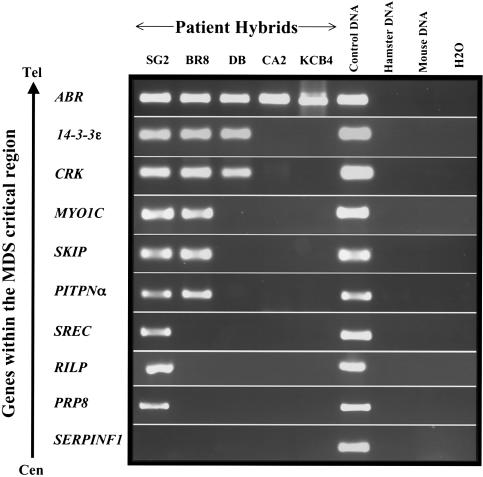
Deletions of 17p13.3, including the LIS1 gene, result in the brain malformation lissencephaly, which is characterized by reduced gyration and cortical thickening; however, the phenotype can vary from isolated lissencephaly sequence (ILS) to Miller-Dieker syndrome (MDS). At the clinical level, these two phenotypes can be differentiated by the presence of significant dysmorphic facial features and a more severe grade of lissencephaly in MDS. Previous work has suggested that children with MDS have a larger deletion than those with ILS, but the precise boundaries of the MDS critical region and causative genes other than LIS1 have never been fully determined. We have completed a physical and transcriptional map of the 17p13.3 region from LIS1 to the telomere. Using fluorescence in situ hybridization, we have mapped the deletion size in 19 children with ILS, 11 children with MDS, and 4 children with 17p13.3 deletions not involving LIS1. We show that the critical region that differentiates ILS from MDS at the molecular level can be reduced to 400 kb. Using somatic cell hybrids from selected patients, we have identified eight genes that are consistently deleted in patients classified as having MDS. In addition, deletion of the genes CRK and 14-3-3 epsilon delineates patients with the most severe lissencephaly grade. On the basis of recent functional data and the creation of a mouse model suggesting a role for 14-3-3 epsilon in cortical development, we suggest that deletion of one or both of these genes in combination with deletion of LIS1 may contribute to the more severe form of lissencephaly seen only in patients with MDS.
Figures

Similar articles
-
A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3.Hum Mol Genet. 1997 Feb;6(2):147-55. doi: 10.1093/hmg/6.2.147. Hum Mol Genet. 1997. PMID: 9063734
-
Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome.Hum Mol Genet. 1997 Feb;6(2):157-64. doi: 10.1093/hmg/6.2.157. Hum Mol Genet. 1997. PMID: 9063735
-
Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13.JAMA. 1993 Dec 15;270(23):2838-42. doi: 10.1001/jama.270.23.2838. JAMA. 1993. PMID: 7907669 Review.
-
The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene.Hum Mol Genet. 2000 Dec 12;9(20):3019-28. doi: 10.1093/hmg/9.20.3019. Hum Mol Genet. 2000. PMID: 11115846
-
Murine modelling of classical lissencephaly.Neurogenetics. 1999 Apr;2(2):77-86. doi: 10.1007/s100480050056. Neurogenetics. 1999. PMID: 10369882 Review.
Cited by
-
Cellular and clinical impact of haploinsufficiency for genes involved in ATR signaling.Am J Hum Genet. 2007 Jul;81(1):77-86. doi: 10.1086/518696. Epub 2007 May 17. Am J Hum Genet. 2007. PMID: 17564965 Free PMC article.
-
Genetics and biology of microcephaly and lissencephaly.Semin Pediatr Neurol. 2009 Sep;16(3):120-6. doi: 10.1016/j.spen.2009.07.001. Semin Pediatr Neurol. 2009. PMID: 19778709 Free PMC article. Review.
-
Malformations of Cerebral Cortex Development: Molecules and Mechanisms.Annu Rev Pathol. 2019 Jan 24;14:293-318. doi: 10.1146/annurev-pathmechdis-012418-012927. Annu Rev Pathol. 2019. PMID: 30677308 Free PMC article. Review.
-
Haploinsufficiency of DNA Damage Response Genes and their Potential Influence in Human Genomic Disorders.Curr Genomics. 2008 May;9(3):137-46. doi: 10.2174/138920208784340795. Curr Genomics. 2008. PMID: 19440510 Free PMC article.
-
Organoids as preclinical models of human disease: progress and applications.Med Rev (2021). 2024 Mar 14;4(2):129-153. doi: 10.1515/mr-2023-0047. eCollection 2024 Apr. Med Rev (2021). 2024. PMID: 38680680 Free PMC article. Review.
References
Electronic-Database Information
-
- CEPH-Généthon Database, http://www.cephb.fr/
-
- GenBank, http://www.ncbi.nlm.nih.gov/entrez/, (for RPCI4-765O05 [accession no. AL137038], RPCI5-59D14 [accession no. AC006435], RPCI5-1029F21 [accession no. AC015853], RPCI5-1037N22 [accession no. AL450226], RPCI5-1127L24 [accession no. AC008087], RPCI11-4F24 [accession no. AC007873], RPCI11-22G12 [accession no. AC016292], RPCI11-74E22 [accession no. AC005696], RPCI11-107N19 [accession no. AC006405], RPCI11-135N5 [accession no. AC015799], RPCI11-216P6 [accession no. AC015884], RPCI11-356I18 [accession no. AC036164], RPCI11-380H7 [accession no. AC021705], RPCI11-411G7 [accession no. AC027455], RPCI11-433M14 [accession no. AC068936], RPCI11-667K14 [accession no. AC090617], RPCI11-676J12 [accession no. AC087392], RPCI11-818O24 [accession no. AC032044], CTB-11O23 [accession no. AC002316], BAC407I21 [accession no. AF322450], CTB-145P4 [accession no. AC002093], CTD-2326F1 [accession no. AC109339], CTD-2348K1 [accession no. AC108004], CTD-2507J6 [accession no. AC107911], CTD-2545H1 [accession no. AC099684], CTD-2573J8 [accession no. AC108006], CTD-3086F13 [accession no. AC099721] and yRM2111 (17pTEL) [accession no. AF240580], RPCI23-78H4 [accession no. AL669897], RPCI23-190H20 [accession no. AL663094], RPCI11-305G1 [accession no. AC032038], RPCI11-713H12 [accession no. AC025518], NUDEL [accession no. NM_030808], profilin1 [accession no. NM_005022], KIAA0664 [accession no. XM_03478], LIS1 [accession no. XM_034770], MNT [accession no. XM_012601], SRR [accession no. NM_021947], KIAA0397 [accession no. AB007857], C17orf31 [or KIAA0732] [accession no. XM_029470], KIAA1401 [accession no. AB037822], HIC1 [accession no. NM_006497], DPH2L [accession no. NM_001383], RPA1 [accession no. NM_002945], SERPINF2 [accession no. NM_000934], SERPINF1 [accession no. NM_002615], KIAA1936 [accession no. XM_056082], PRP8 [accession no. XM_028335], RILP [accession no. NM_031430], SREC [accession no. XM_008489], PITPN-α [accession no. NM_006224], SKIP [accession no. NM_016532], MYO1C [accession no. XM_028385], CRK III [accession no. NM_005206 and NM_016823], 14-3-3ε [accession no. NM_006761], ABR [accession no. NM_021962 and NM_001092], TIMM22 [accession no. XM_033715], NXN [accession no. XM_033714], HC90 [accession no. AF177344], CGI-150 [accession no. NM_016080], ELP [accession no. AF229804], HC56 [accession no. AF177341], FLJ22282 [accession no. NM_024792], FLJ32270 [accession no. AK056832], FLJ10979 [accession no. NM_018289], FLJ31761 [accession no. AK056323], RPH3AL [accession no. NM_006987], and DOC2B [accession no. NM_003585])
-
- HUGO chromosome 17, http://gdbwww.gdb.org/hugo/chr17/
-
- Lissencephaly Network, http://www.lissencephaly.org
-
- NCBI UniGene, http://www.ncbi.nlm.nih.gov/UniGene/
References
-
- Adachi H, Tsujimoto M (2002) Characterization of human gene encoding scavenger receptor expressed by endothelial cells (SREC) and its regulation by a novel transcription factor, endothelial zinc finger protein-2 (EZF-2). J Biol Chem 277:24014–24021 - PubMed
-
- Cardoso C, Leventer RJ, Matsumoto N, Kuc JA, Ramocki MB, Mewborn SK, Dudlicek LL, May LF, Mills PL, Das S, Pilz DT, Dobyns WB, Ledbetter DH (2000) The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum Mol Genet 9:3019–3028 - PubMed
-
- Chong SS, Tanigami A, Roschke AV, Ledbetter DH (1996) 14-3-3 epsilon has no homology to LIS1 and lies telomeric to it on chromosome 17p13.3 outside the Miller-Dieker syndrome chromosome region. Genome Res 6:735–741 - PubMed
-
- Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith AC, Dobyns WB, Ledbetter DH. (1997) A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet 6:147–155 - PubMed
-
- Christian, SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH (1999) Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11–q13). Hum Mol Genet 8:1025–1037 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
