

Rapidly Progressive Dementia
- PMID: 27042906
- PMCID: PMC4879977
- DOI: 10.1212/CON.0000000000000319
Rapidly Progressive Dementia
Abstract
Purpose of review: This article presents a practical and informative approach to the evaluation of a patient with a rapidly progressive dementia (RPD).
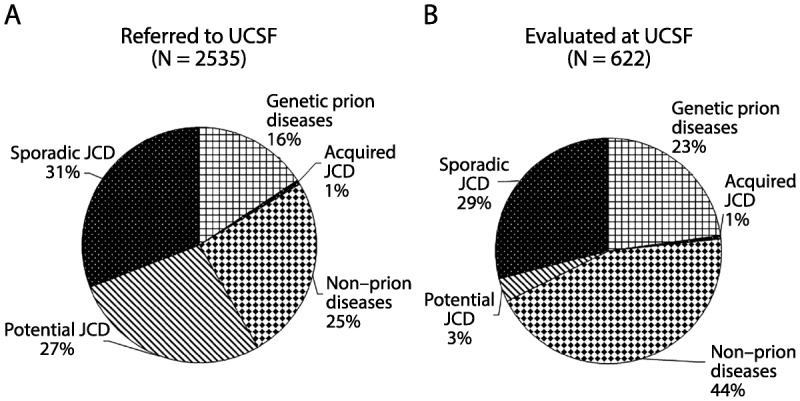
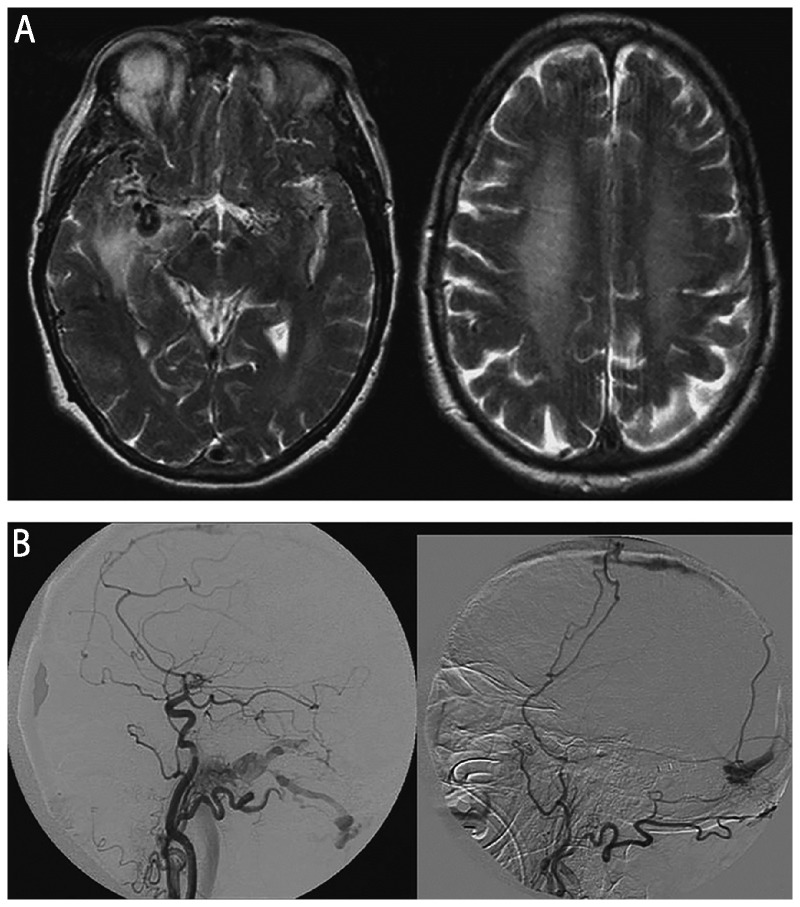
Recent findings: Prion diseases are the prototypical causes of RPD, but reversible causes of RPD might mimic prion disease and should always be considered in a differential diagnosis. Aside from prion diseases, the most common causes of RPD are atypical presentations of other neurodegenerative disorders, curable disorders including autoimmune encephalopathies, as well as some infections, and neoplasms. Numerous recent case reports suggest dural arterial venous fistulas sometimes cause RPDs.
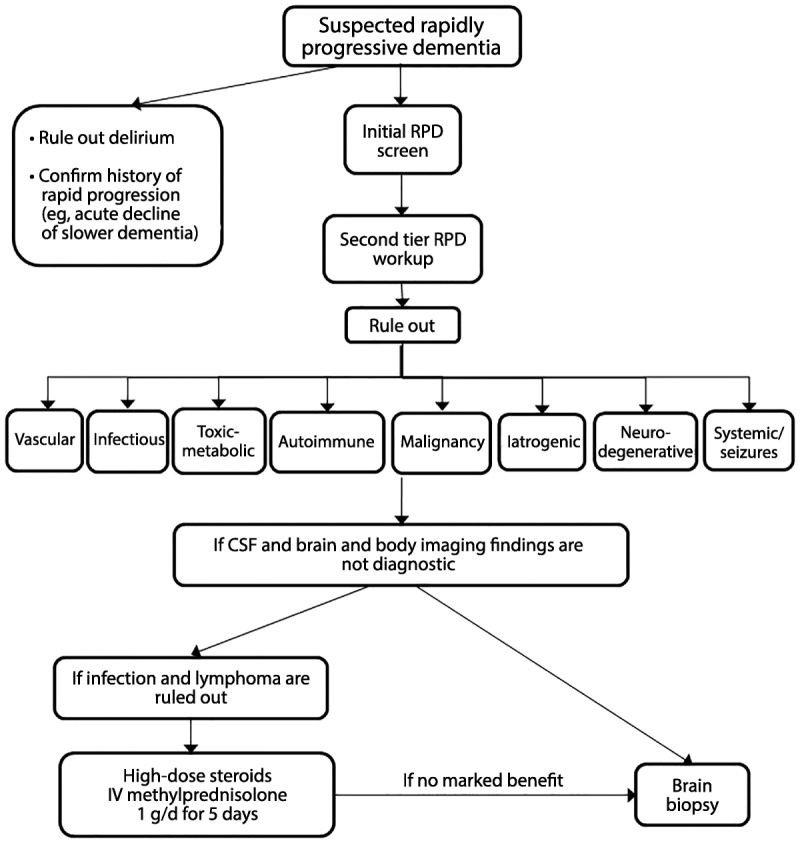
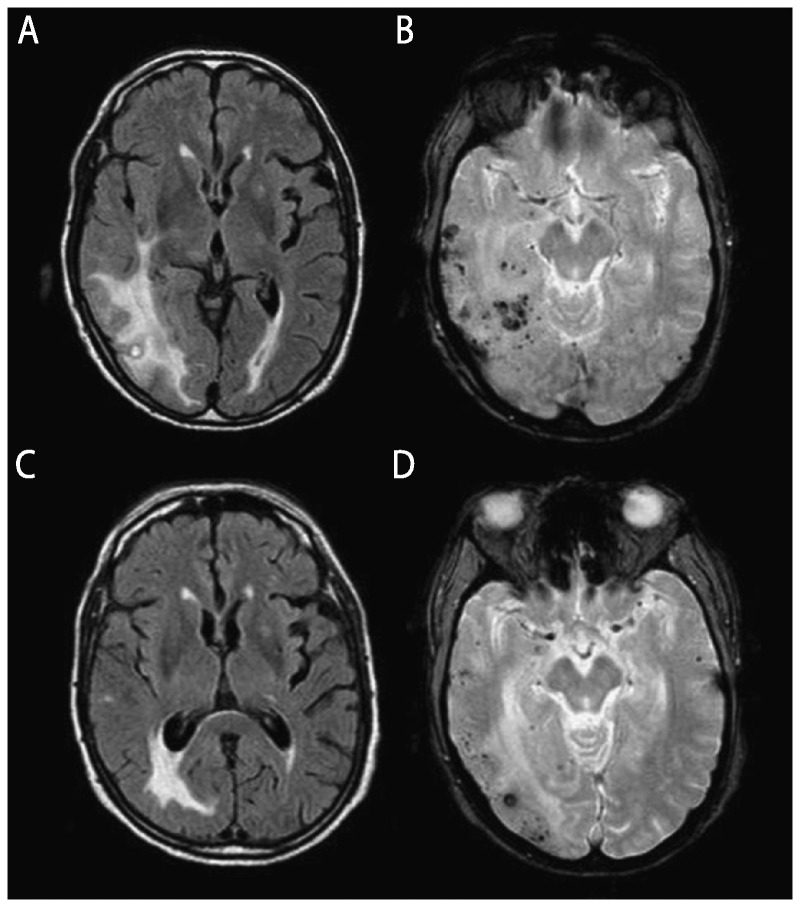
Summary: RPDs, in which patients typically develop dementia over weeks to months, require an alternative differential than the slowly progressive dementias that occur over a few years. Because of their rapid decline, patients with RPDs necessitate urgent evaluation and often require an extensive workup, typically with multiple tests being sent or performed concurrently. Jakob-Creutzfeldt disease, perhaps the prototypical RPD, is often the first diagnosis many neurologists consider when treating a patient with rapid cognitive decline. Many conditions other than prion disease, however, including numerous reversible or curable conditions, can present as an RPD. This chapter discusses some of the major etiologies for RPDs and offers an algorithm for diagnosis.
Figures
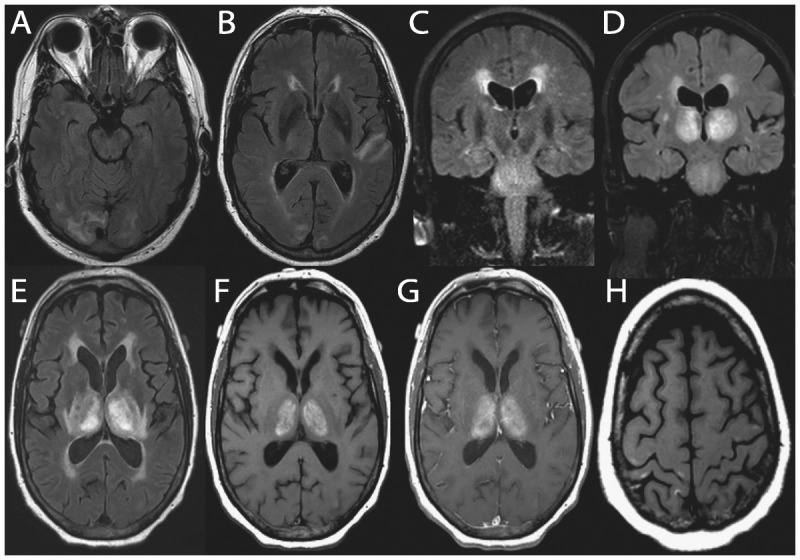
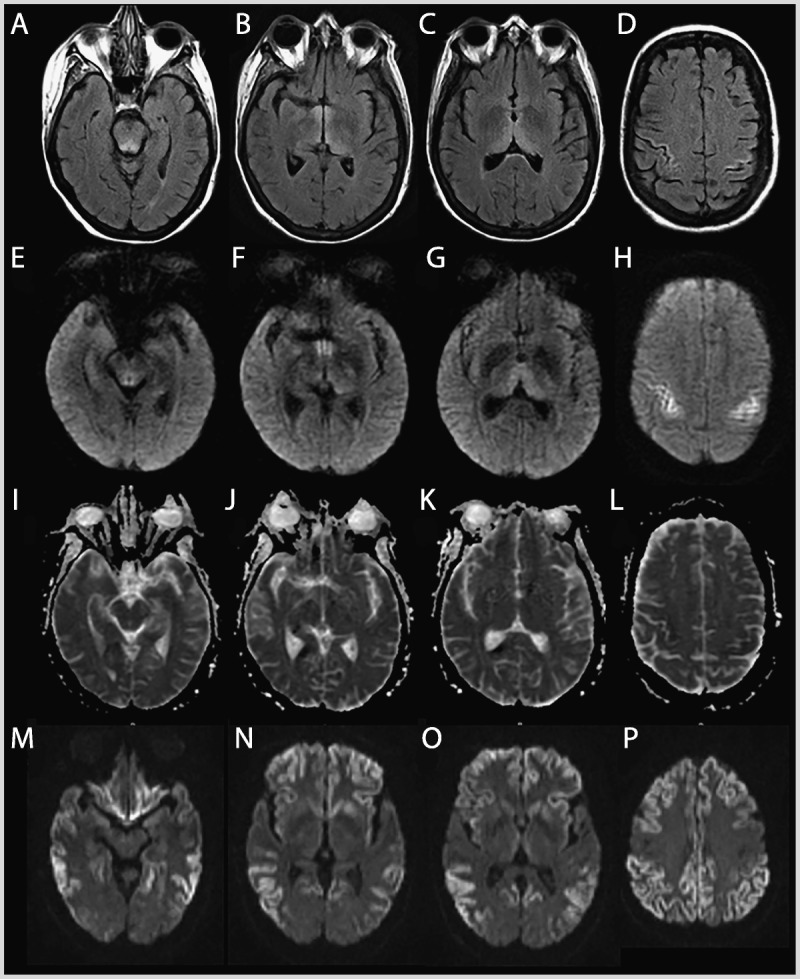
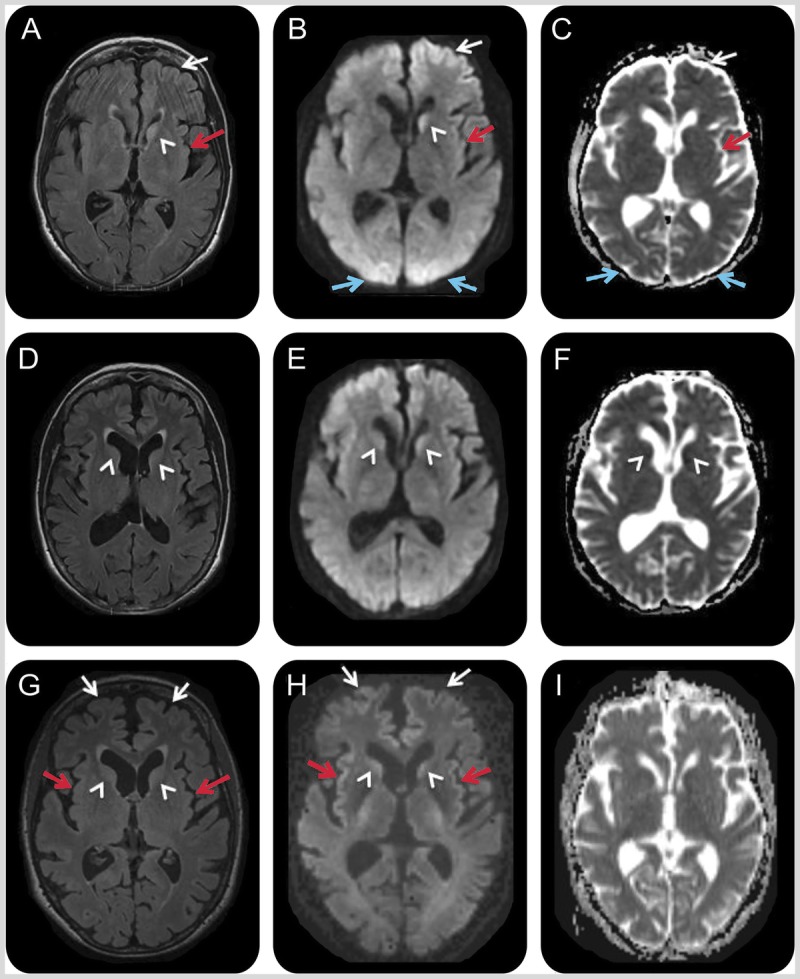
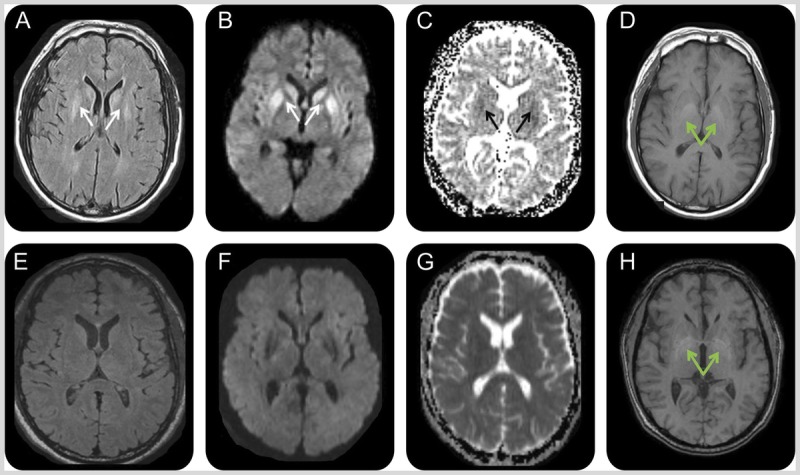
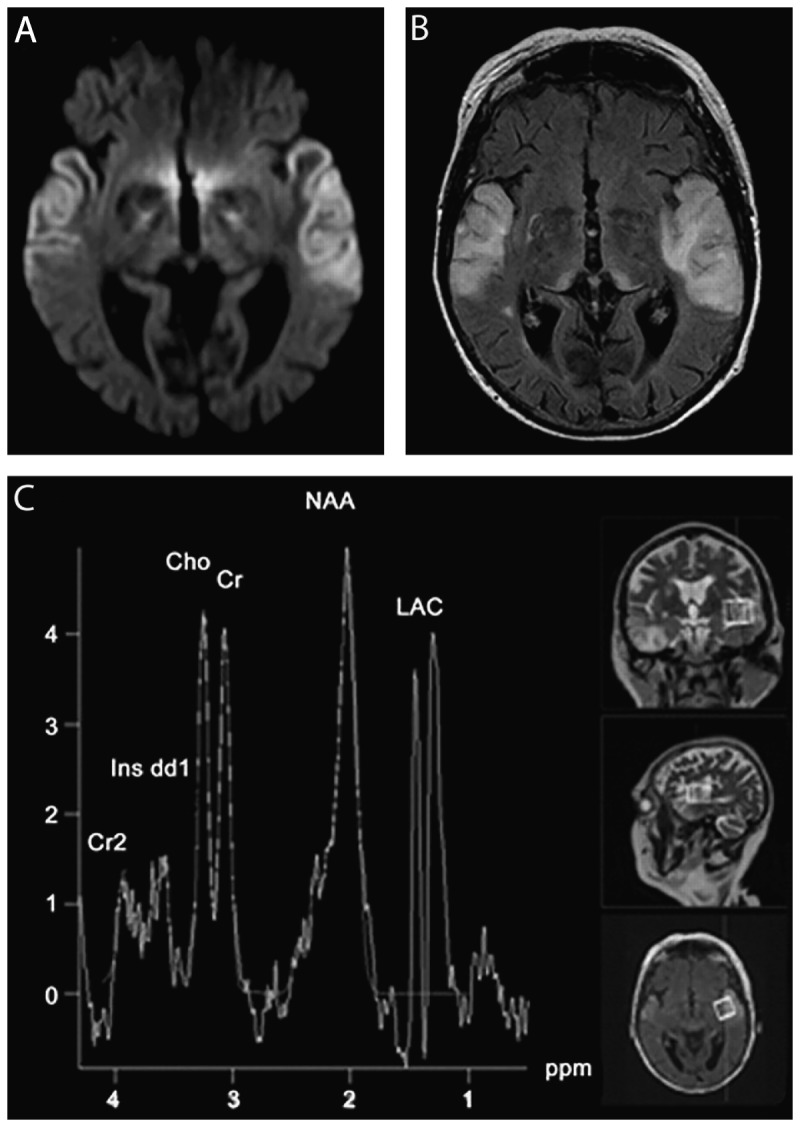
Similar articles
-
Rapidly progressive dementia: prion diseases and other rapid dementias.Continuum (Minneap Minn). 2010 Apr;16(2 Dementia):31-56. doi: 10.1212/01.CON.0000368211.79211.4c. Continuum (Minneap Minn). 2010. PMID: 22810280
-
Differential diagnosis with other rapid progressive dementias in human prion diseases.Handb Clin Neurol. 2018;153:371-397. doi: 10.1016/B978-0-444-63945-5.00020-9. Handb Clin Neurol. 2018. PMID: 29887146 Review.
-
Rapidly progressive young-onset dementias: neuropsychiatric aspects.Psychiatr Clin North Am. 2015 Jun;38(2):221-32. doi: 10.1016/j.psc.2015.01.001. Epub 2015 Mar 2. Psychiatr Clin North Am. 2015. PMID: 25998112 Review.
-
Rapidly progressive dementias - aetiologies, diagnosis and management.Nat Rev Neurol. 2022 Jun;18(6):363-376. doi: 10.1038/s41582-022-00659-0. Epub 2022 May 4. Nat Rev Neurol. 2022. PMID: 35508635 Free PMC article. Review.
-
Rapidly progressive dementias and the treatment of human prion diseases.Expert Opin Pharmacother. 2011 Jan;12(1):1-12. doi: 10.1517/14656566.2010.514903. Epub 2010 Nov 23. Expert Opin Pharmacother. 2011. PMID: 21091283 Free PMC article. Review.
Cited by
-
Pseudotumoral Presentation of Cerebral Amyloid-Beta Angiopathy: Case Report and Review of Literature.Psychiatry Investig. 2021 Jun;18(6):479-485. doi: 10.30773/pi.2020.0201. Epub 2021 Jun 17. Psychiatry Investig. 2021. PMID: 34130443 Free PMC article.
-
Diagnosis and Management of Dementia: Review.JAMA. 2019 Oct 22;322(16):1589-1599. doi: 10.1001/jama.2019.4782. JAMA. 2019. PMID: 31638686 Free PMC article. Review.
-
An Evaluation of Rapidly Progressive Dementia Culminating in a Diagnosis of Creutzfeldt-Jakob Disease.Case Rep Infect Dis. 2018 Sep 23;2018:2374179. doi: 10.1155/2018/2374179. eCollection 2018. Case Rep Infect Dis. 2018. PMID: 30345127 Free PMC article.
-
Subjective cognitive decline, mild cognitive impairment, and dementia - syndromic approach: recommendations of the Scientific Department of Cognitive Neurology and Aging of the Brazilian Academy of Neurology.Dement Neuropsychol. 2022 Nov 28;16(3 Suppl 1):1-24. doi: 10.1590/1980-5764-DN-2022-S101PT. eCollection 2022 Sep. Dement Neuropsychol. 2022. PMID: 36533160 Free PMC article.
-
Rapidly progressive dementia due to intravascular lymphoma: A prion disease reference center experience.Eur J Neurol. 2024 Jan;31(1):e16068. doi: 10.1111/ene.16068. Epub 2023 Sep 22. Eur J Neurol. 2024. PMID: 37738529 Free PMC article.
References
-
- Gibbs CJ., Jr Spongiform encephalopathies—slow, latent, and temperate virus infections—in retrospect. In: Prusiner SB, Collinge J, Powell J, et al. eds. Prion diseases of humans and animals. London, UK: Ellis Horwood, 1992: 53– 62.
-
- Katscher F. It’s Jakob’s disease, not Creutzfeldt’s. Nature 1998; 393(6680): 11 doi:10.1038/29862. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
