

MYBPC3 D389V Variant Induces Hypercontractility in Cardiac Organoids
- PMID: 39594661
- PMCID: PMC11592734
- DOI: 10.3390/cells13221913
MYBPC3 D389V Variant Induces Hypercontractility in Cardiac Organoids
Abstract
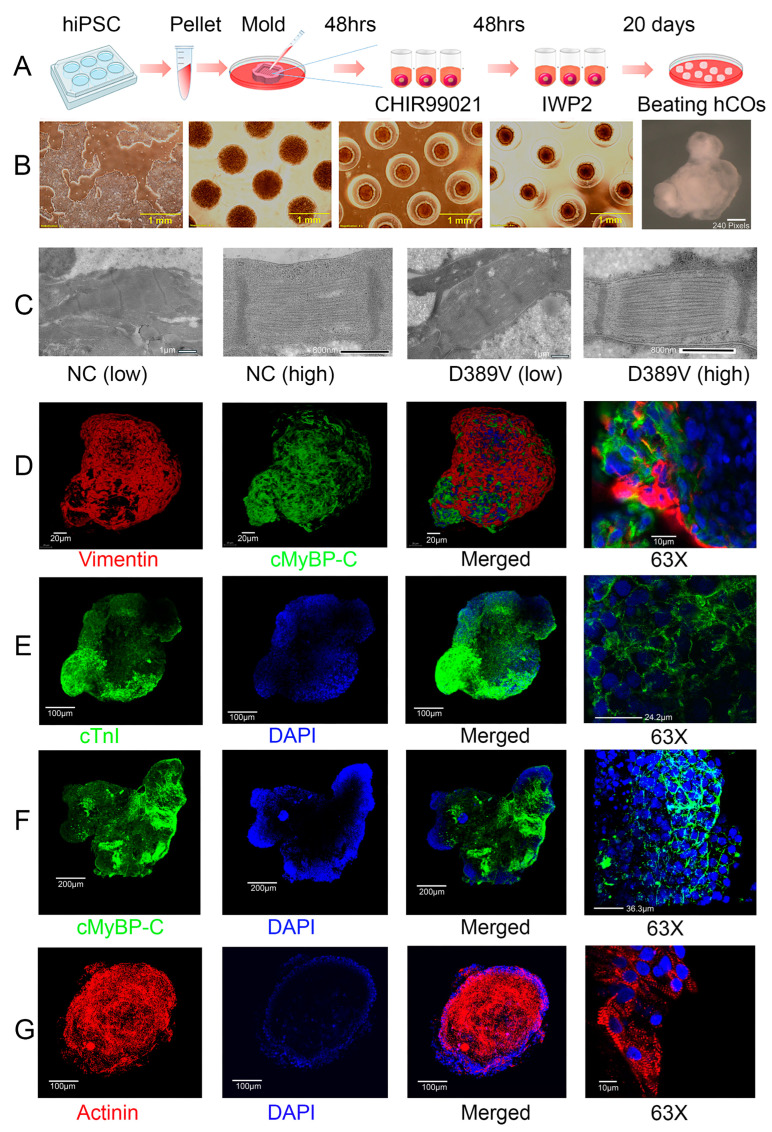
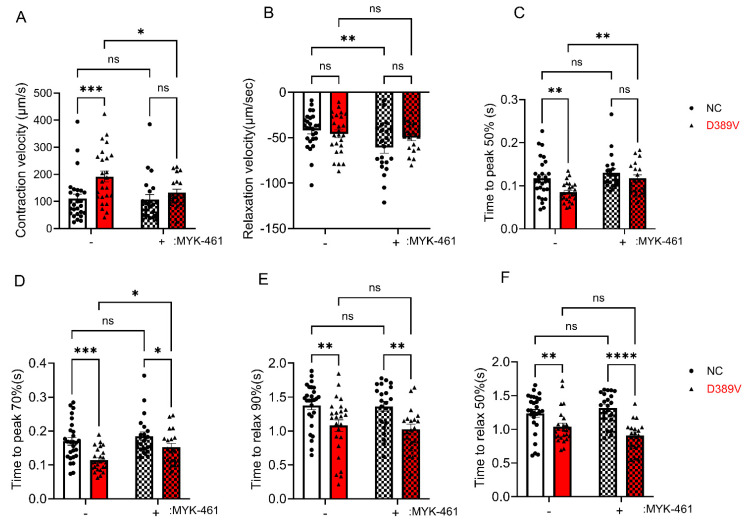
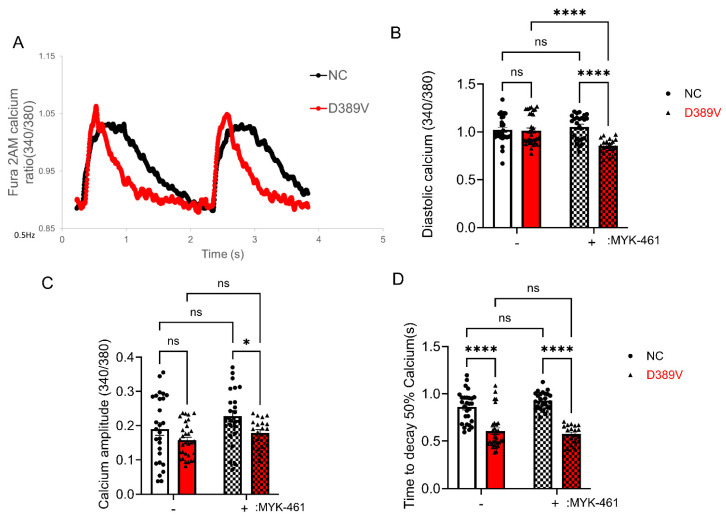
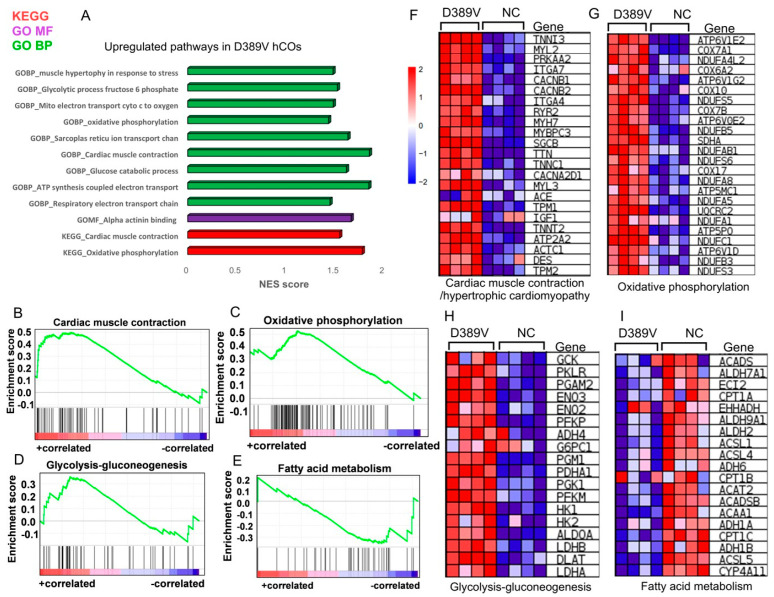
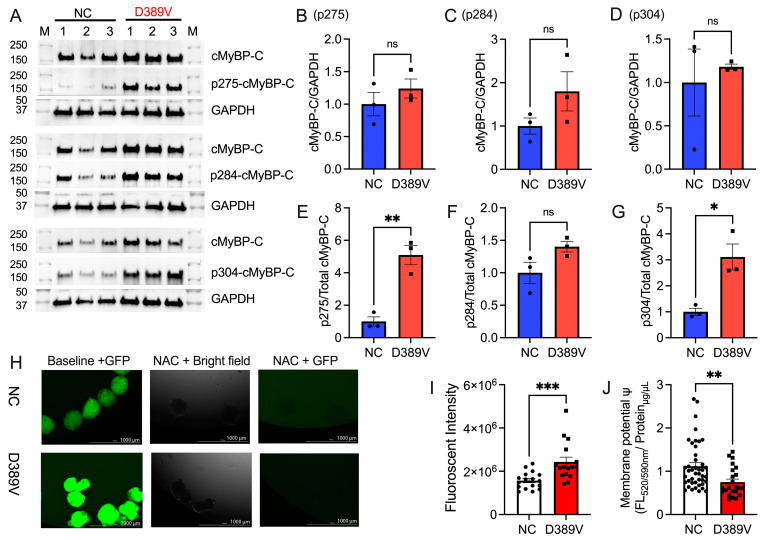
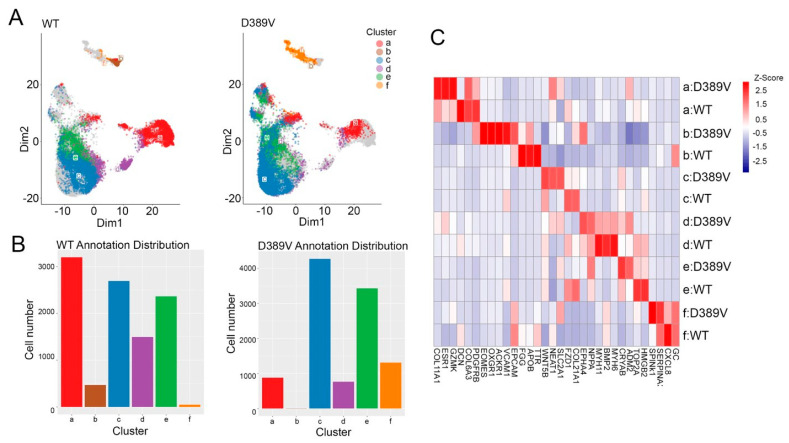
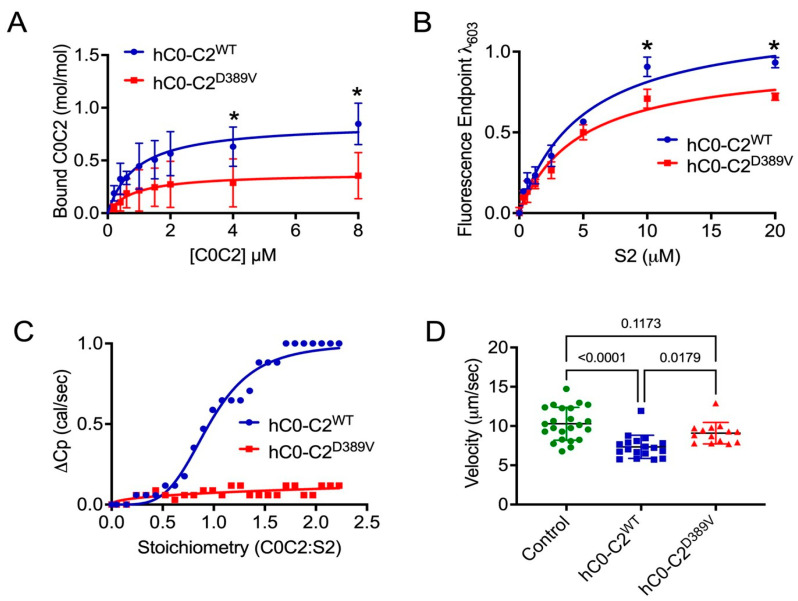
MYBPC3, encoding cardiac myosin binding protein-C (cMyBP-C), is the most mutated gene known to cause hypertrophic cardiomyopathy (HCM). However, since little is known about the underlying etiology, additional in vitro studies are crucial to defining the underlying molecular mechanisms. Accordingly, this study aimed to investigate the molecular mechanisms underlying the pathogenesis of HCM associated with a polymorphic variant (D389V) in MYBPC3 by using isogenic human-induced pluripotent stem cell (hiPSC)-derived cardiac organoids (hCOs). The hiPSC-derived cardiomyocytes (hiPSC-CMs) and hCOs were generated from human subjects to define the molecular, cellular, functional, and energetic changes caused by the MYBPC3D389V variant, which is associated with increased fractional shortening and highly prevalent in South Asian descendants. Recombinant C0-C2, N' region of cMyBP-C (wild-type and D389V), and myosin S2 proteins were also utilized to perform binding and motility assays in vitro. Confocal and electron microscopic analyses of hCOs generated from noncarriers (NC) and carriers of the MYBPC3D389V variant revealed the presence of highly organized sarcomeres. Furthermore, functional experiments showed hypercontractility, faster calcium cycling, and faster contractile kinetics in hCOs expressing MYBPC3D389V than NC hCOs. Interestingly, significantly increased cMyBP-C phosphorylation in MYBPC3D389V hCOs was observed, but without changes in total protein levels, in addition to higher oxidative stress and lower mitochondrial membrane potential (ΔΨm). Next, spatial mapping revealed the presence of endothelial cells, fibroblasts, macrophages, immune cells, and cardiomyocytes in the hCOs. The hypercontractile function was significantly improved after the treatment of the myosin inhibitor mavacamten (CAMZYOS®) in MYBPC3D389V hCOs. Lastly, various vitro binding assays revealed a significant loss of affinity in the presence of MYBPC3D389V with myosin S2 region as a likely mechanism for hypercontraction. Conceptually, we showed the feasibility of assessing the functional and molecular mechanisms of HCM using highly translatable hCOs through pragmatic experiments that led to determining the MYBPC3D389V hypercontractile phenotype, which was rescued by the administration of a myosin inhibitor.
Keywords: MYBPC3; cardiac organoids; hypercontraction; hypertrophic cardiomyopathy; mavacamten.
Conflict of interest statement
Sadayappan provides consulting and collaborative research studies to the Leducq Foundation (CURE-PLAN), Red Saree Inc., Alexion, Regel Therapeutics, Affinia Therapeutics Inc., Cardiocare Genetics—Cosmogene Skincare Pvt Ltd., but such work is unrelated to the content of this article.
Figures
Update of
-
MYBPC3 D389V Variant Induces Hypercontractility in Cardiac Organoids.bioRxiv [Preprint]. 2024 May 30:2024.05.29.596463. doi: 10.1101/2024.05.29.596463. bioRxiv. 2024. Update in: Cells. 2024 Nov 19;13(22):1913. doi: 10.3390/cells13221913. PMID: 38853909 Free PMC article. Updated. Preprint.
Similar articles
-
MYBPC3 D389V Variant Induces Hypercontractility in Cardiac Organoids.bioRxiv [Preprint]. 2024 May 30:2024.05.29.596463. doi: 10.1101/2024.05.29.596463. bioRxiv. 2024. Update in: Cells. 2024 Nov 19;13(22):1913. doi: 10.3390/cells13221913. PMID: 38853909 Free PMC article. Updated. Preprint.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Hypertrophic cardiomyopathy β-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state.Proc Natl Acad Sci U S A. 2021 Jun 15;118(24):e2025030118. doi: 10.1073/pnas.2025030118. Proc Natl Acad Sci U S A. 2021. PMID: 34117120 Free PMC article.
-
The HCM-linked W792R mutation in cardiac myosin-binding protein C reduces C6 FnIII domain stability.Am J Physiol Heart Circ Physiol. 2018 Jun 1;314(6):H1179-H1191. doi: 10.1152/ajpheart.00686.2017. Epub 2018 Feb 16. Am J Physiol Heart Circ Physiol. 2018. PMID: 29451820 Free PMC article.
-
Trends in Surgical and Nonsurgical Aesthetic Procedures: A 14-Year Analysis of the International Society of Aesthetic Plastic Surgery-ISAPS.Aesthetic Plast Surg. 2024 Oct;48(20):4217-4227. doi: 10.1007/s00266-024-04260-2. Epub 2024 Aug 5. Aesthetic Plast Surg. 2024. PMID: 39103642 Review.
References
-
- Kramer C.M., Appelbaum E., Desai M.Y., Desvigne-Nickens P., DiMarco J.P., Friedrich M.G., Geller N., Heckler S., Ho C.Y., Jerosch-Herold M., et al. Hypertrophic Cardiomyopathy Registry: The rationale and design of an international, observational study of hypertrophic cardiomyopathy. Am. Heart J. 2015;170:223–230. doi: 10.1016/j.ahj.2015.05.013. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
