

Innovations in the Treatment of Dystrophic Epidermolysis Bullosa (DEB): Current Landscape and Prospects
- PMID: 37337559
- PMCID: PMC10277004
- DOI: 10.2147/TCRM.S386923
Innovations in the Treatment of Dystrophic Epidermolysis Bullosa (DEB): Current Landscape and Prospects
Abstract
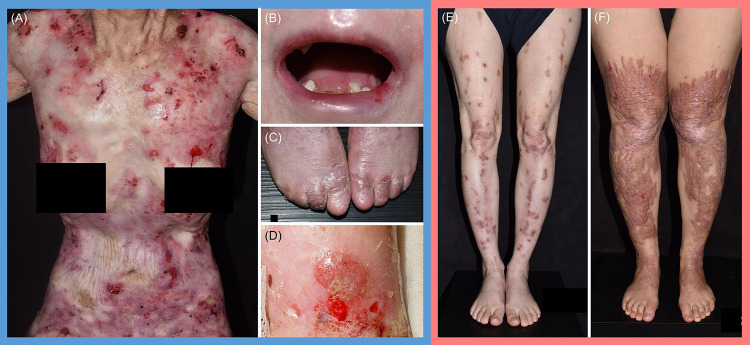
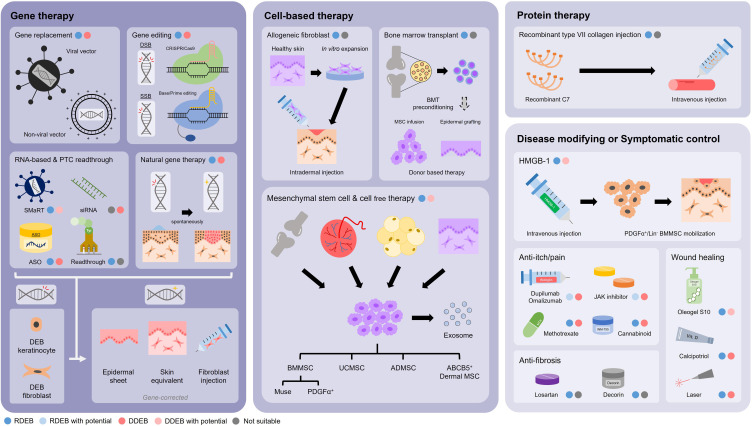
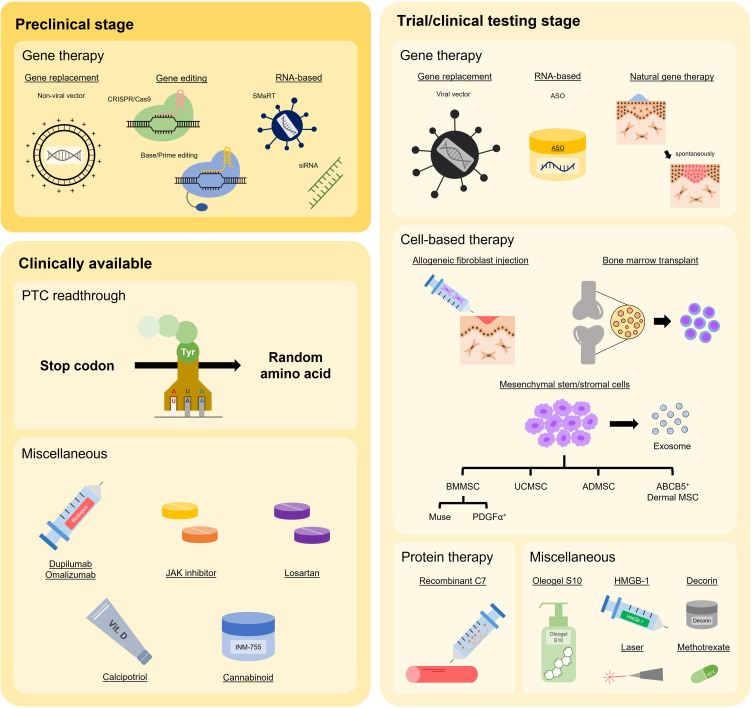
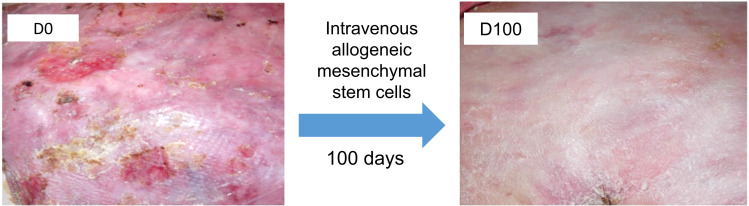
Dystrophic epidermolysis bullosa (DEB) is one of the major types of EB, a rare hereditary group of trauma-induced blistering skin disorders. DEB is caused by inherited pathogenic variants in the COL7A1 gene, which encodes type VII collagen, the major component of anchoring fibrils which maintain adhesion between the outer epidermis and underlying dermis. DEB can be subclassified into dominant (DDEB) and recessive (RDEB) forms. Generally, DDEB has a milder phenotype, while RDEB patients often have more extensive blistering, chronic inflammation, skin fibrosis, and a propensity for squamous cell carcinoma development, collectively impacting on daily activities and life expectancy. At present, best practice treatments are mostly supportive, and thus there is a considerable burden of disease with unmet therapeutic need. Over the last 20 years, considerable translational research efforts have focused on either trying to cure DEB by direct correction of the COL7A1 gene pathology, or by modifying secondary inflammation to lessen phenotypic severity and improve patient symptoms such as poor wound healing, itch, and pain. In this review, we provide an overview and update on various therapeutic innovations for DEB, including gene therapy, cell-based therapy, protein therapy, and disease-modifying and symptomatic control agents. We outline the progress and challenges for each treatment modality and identify likely prospects for future clinical impact.
Keywords: blister; cell therapy; fibrosis; gene therapy; inflammation; skin.
© 2023 Hou et al.
Conflict of interest statement
The authors report no conflicts of interest in this work.
Figures

Similar articles
-
Clinical characteristics, healthcare use, and annual costs among patients with dystrophic epidermolysis bullosa.Orphanet J Rare Dis. 2022 Sep 29;17(1):367. doi: 10.1186/s13023-022-02509-0. Orphanet J Rare Dis. 2022. PMID: 36175960 Free PMC article.
-
QR-313, an Antisense Oligonucleotide, Shows Therapeutic Efficacy for Treatment of Dominant and Recessive Dystrophic Epidermolysis Bullosa: A Preclinical Study.J Invest Dermatol. 2021 Apr;141(4):883-893.e6. doi: 10.1016/j.jid.2020.08.018. Epub 2020 Sep 16. J Invest Dermatol. 2021. PMID: 32946877
-
The clinical spectrum of dystrophic epidermolysis bullosa.Br J Dermatol. 2002 Feb;146(2):267-74. doi: 10.1046/j.1365-2133.2002.04607.x. Br J Dermatol. 2002. PMID: 11903238
-
Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine novel COL7A1 variants.J Dermatol Sci. 2007 Jun;46(3):169-78. doi: 10.1016/j.jdermsci.2007.02.006. Epub 2007 Apr 10. J Dermatol Sci. 2007. PMID: 17425959 Review.
-
Gene therapy: pursuing restoration of dermal adhesion in recessive dystrophic epidermolysis bullosa.Exp Dermatol. 2014 Jan;23(1):1-6. doi: 10.1111/exd.12246. Exp Dermatol. 2014. PMID: 24107073 Review.
Cited by
-
Management of Skin Lesions in Patients with Epidermolysis Bullosa by Topical Treatment: Systematic Review and Meta-Analysis.Healthcare (Basel). 2024 Jan 19;12(2):261. doi: 10.3390/healthcare12020261. Healthcare (Basel). 2024. PMID: 38275540 Free PMC article. Review.
-
Clinical Characteristics and Outcomes of Patients Hospitalized with Epidermolysis Bullosa: A Retrospective Population-Based Observational Study in Spain (2016-2021).Biomedicines. 2023 Sep 20;11(9):2584. doi: 10.3390/biomedicines11092584. Biomedicines. 2023. PMID: 37761025 Free PMC article.
-
Genetic Implications and Management of Epidermolysis Bullosa in the Saudi Arabian Population.Cureus. 2024 Aug 12;16(8):e66678. doi: 10.7759/cureus.66678. eCollection 2024 Aug. Cureus. 2024. PMID: 39262533 Free PMC article. Review.
-
"Quality of Life in Epidermolysis Bullosa" and "Epidermolysis Bullosa Burden of Disease": Italian translation, cultural adaptation, and pilot testing of two disease-specific questionnaires.Ital J Pediatr. 2024 Apr 19;50(1):76. doi: 10.1186/s13052-024-01657-2. Ital J Pediatr. 2024. PMID: 38637879 Free PMC article.
-
Intravenous gentamicin therapy induces functional type VII collagen in patients with recessive dystrophic epidermolysis bullosa: an open-label clinical trial.Br J Dermatol. 2024 Jul 16;191(2):267-274. doi: 10.1093/bjd/ljae063. Br J Dermatol. 2024. PMID: 38366625 Free PMC article. Clinical Trial.
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
