

Mechanisms of Resistance to Noncovalent Bruton's Tyrosine Kinase Inhibitors
- PMID: 35196427
- PMCID: PMC9074143
- DOI: 10.1056/NEJMoa2114110
Mechanisms of Resistance to Noncovalent Bruton's Tyrosine Kinase Inhibitors
Abstract
Background: Covalent (irreversible) Bruton's tyrosine kinase (BTK) inhibitors have transformed the treatment of multiple B-cell cancers, especially chronic lymphocytic leukemia (CLL). However, resistance can arise through multiple mechanisms, including acquired mutations in BTK at residue C481, the binding site of covalent BTK inhibitors. Noncovalent (reversible) BTK inhibitors overcome this mechanism and other sources of resistance, but the mechanisms of resistance to these therapies are currently not well understood.
Methods: We performed genomic analyses of pretreatment specimens as well as specimens obtained at the time of disease progression from patients with CLL who had been treated with the noncovalent BTK inhibitor pirtobrutinib. Structural modeling, BTK-binding assays, and cell-based assays were conducted to study mutations that confer resistance to noncovalent BTK inhibitors.
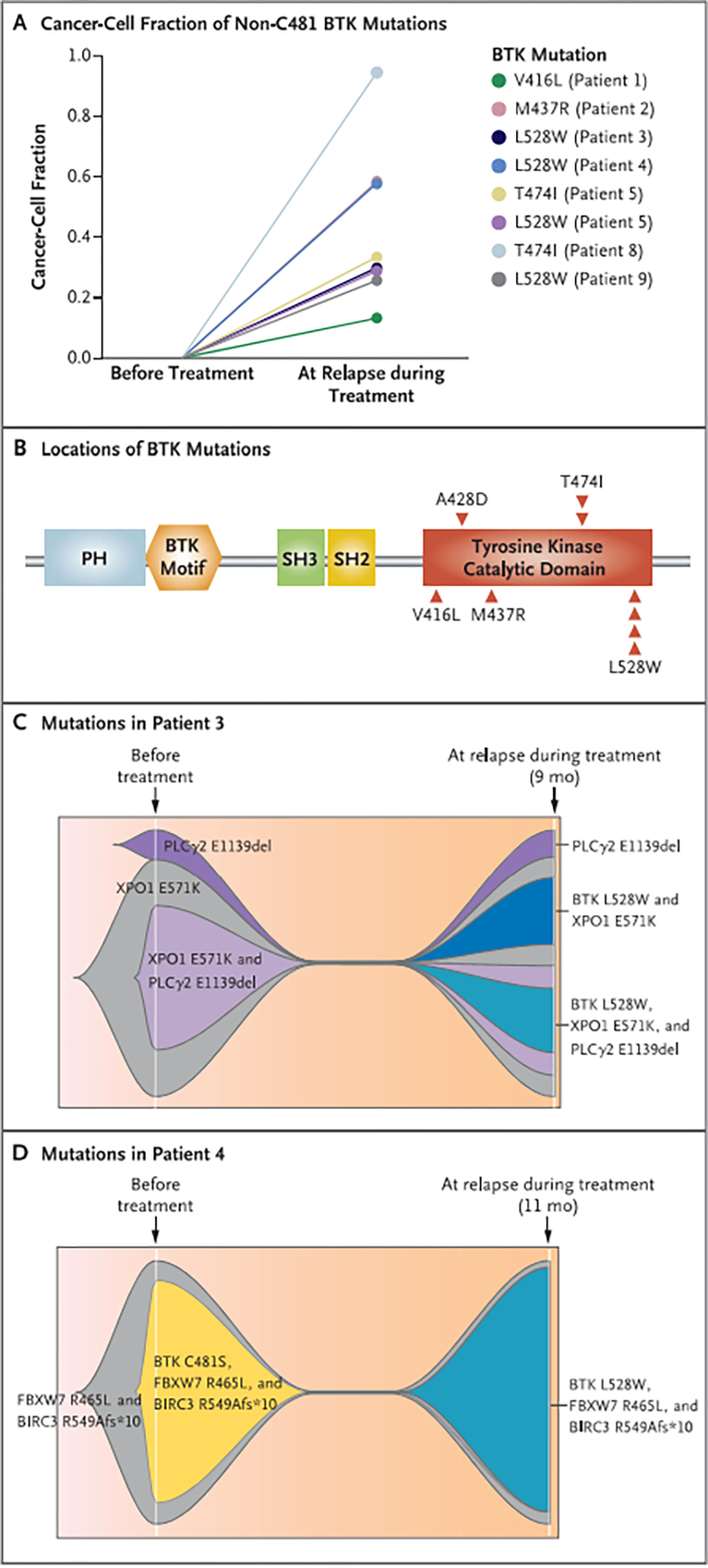
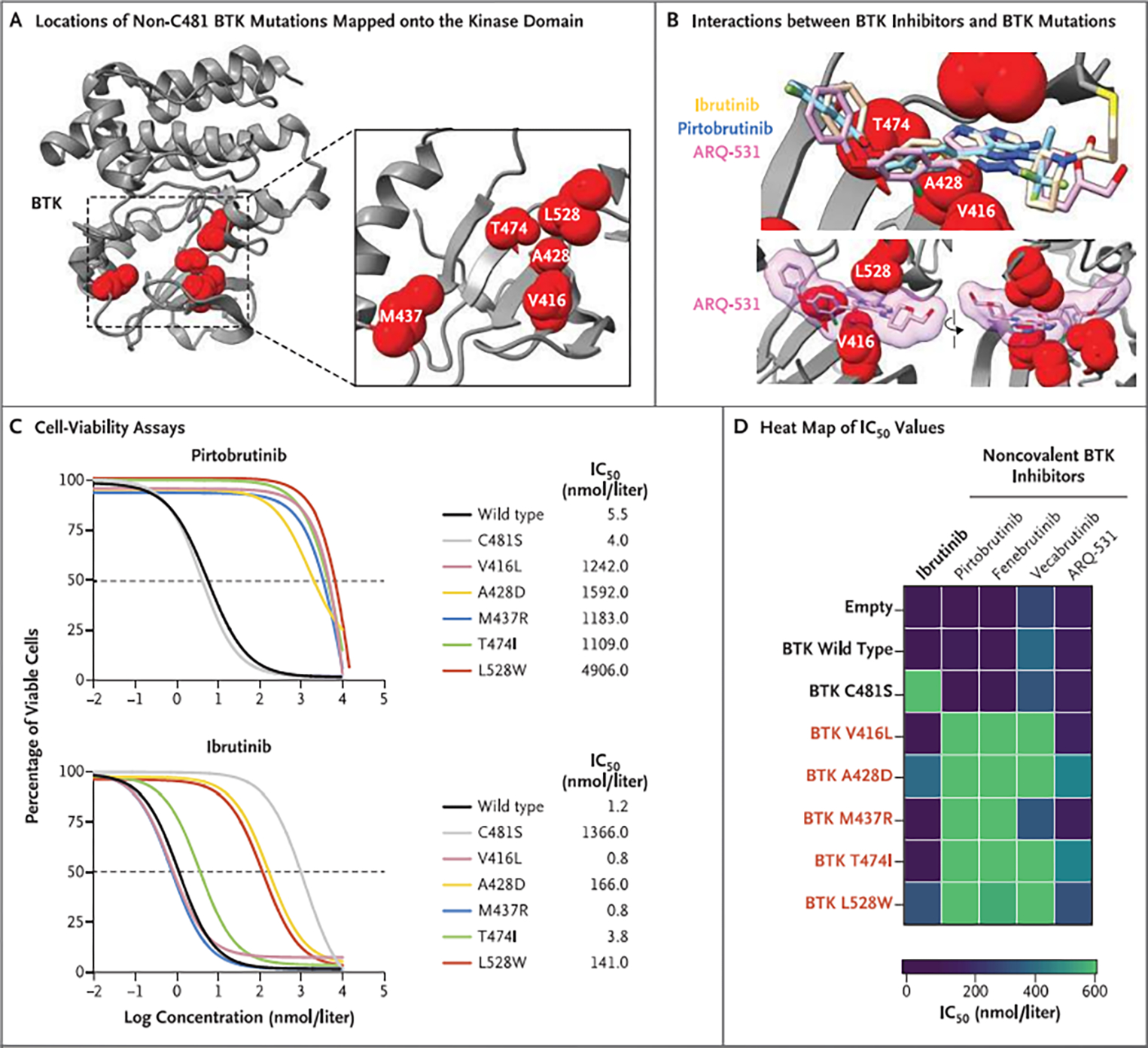
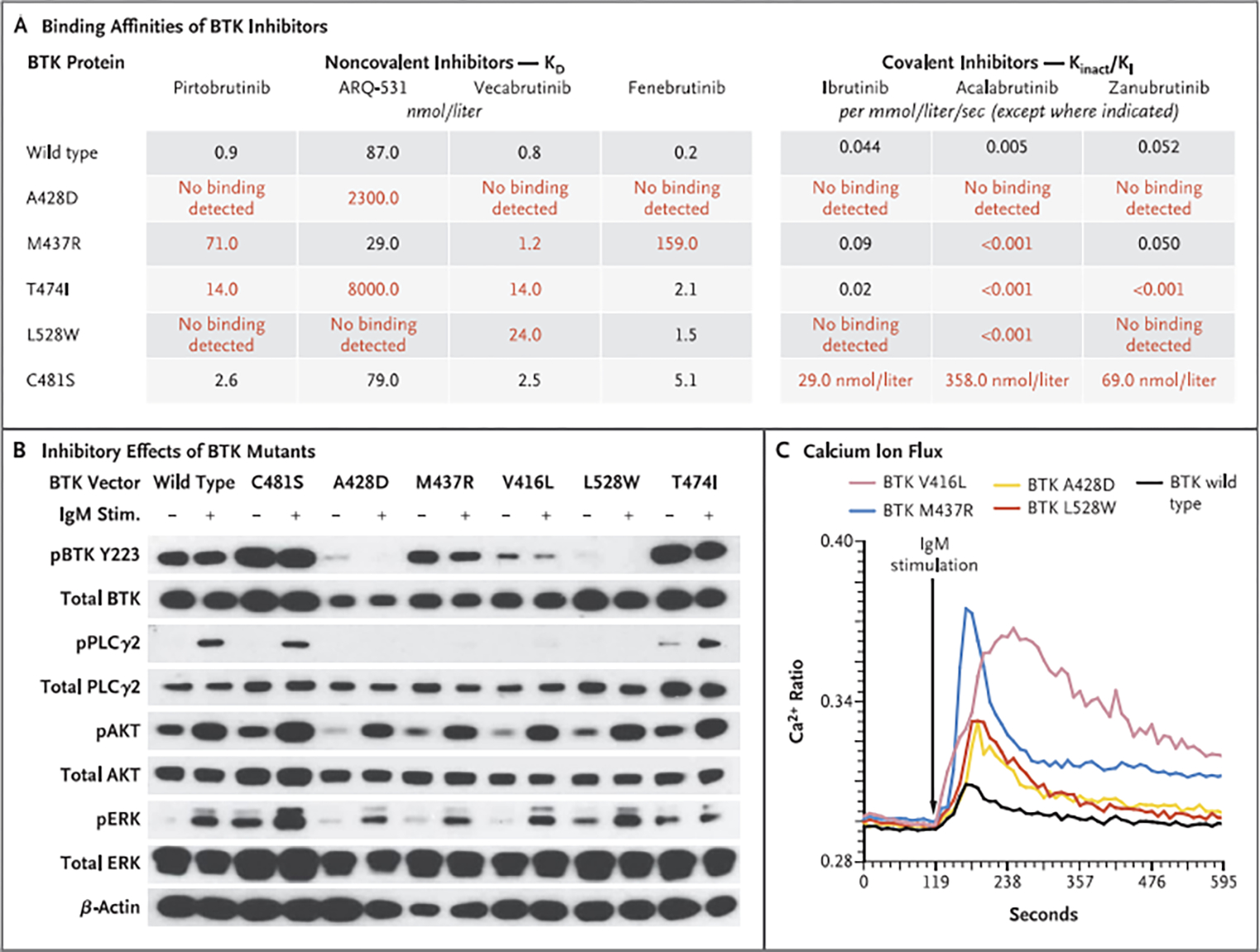
Results: Among 55 treated patients, we identified 9 patients with relapsed or refractory CLL and acquired mechanisms of genetic resistance to pirtobrutinib. We found mutations (V416L, A428D, M437R, T474I, and L528W) that were clustered in the kinase domain of BTK and that conferred resistance to both noncovalent BTK inhibitors and certain covalent BTK inhibitors. Mutations in BTK or phospholipase C gamma 2 (PLCγ2), a signaling molecule and downstream substrate of BTK, were found in all 9 patients. Transcriptional activation reflecting B-cell-receptor signaling persisted despite continued therapy with noncovalent BTK inhibitors.
Conclusions: Resistance to noncovalent BTK inhibitors arose through on-target BTK mutations and downstream PLCγ2 mutations that allowed escape from BTK inhibition. A proportion of these mutations also conferred resistance across clinically approved covalent BTK inhibitors. These data suggested new mechanisms of genomic escape from established covalent and novel noncovalent BTK inhibitors. (Funded by the American Society of Hematology and others.).
Copyright © 2022 Massachusetts Medical Society.
Figures
Comment in
-
Acquired BTK mutations associated with resistance to noncovalent BTK inhibitors.Blood Adv. 2023 Oct 10;7(19):5698-5702. doi: 10.1182/bloodadvances.2022008955. Blood Adv. 2023. PMID: 36661329 Free PMC article. No abstract available.
Similar articles
-
Noncatalytic Bruton's tyrosine kinase activates PLCγ2 variants mediating ibrutinib resistance in human chronic lymphocytic leukemia cells.J Biol Chem. 2020 Apr 24;295(17):5717-5736. doi: 10.1074/jbc.RA119.011946. Epub 2020 Mar 17. J Biol Chem. 2020. PMID: 32184360 Free PMC article.
-
Impact of the clinically approved BTK inhibitors on the conformation of full-length BTK and analysis of the development of BTK resistance mutations in chronic lymphocytic leukemia.Elife. 2024 Dec 27;13:RP95488. doi: 10.7554/eLife.95488. Elife. 2024. PMID: 39728925 Free PMC article.
-
A Review of Resistance Mechanisms to Bruton's Kinase Inhibitors in Chronic Lymphocytic Leukemia.Int J Mol Sci. 2024 May 11;25(10):5246. doi: 10.3390/ijms25105246. Int J Mol Sci. 2024. PMID: 38791284 Free PMC article. Review.
-
Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib.N Engl J Med. 2014 Jun 12;370(24):2286-94. doi: 10.1056/NEJMoa1400029. Epub 2014 May 28. N Engl J Med. 2014. PMID: 24869598 Free PMC article.
-
Resistance Mutations to BTK Inhibitors Originate From the NF-κB but Not From the PI3K-RAS-MAPK Arm of the B Cell Receptor Signaling Pathway.Front Immunol. 2021 Jun 10;12:689472. doi: 10.3389/fimmu.2021.689472. eCollection 2021. Front Immunol. 2021. PMID: 34177947 Free PMC article. Review.
Cited by
-
Acquired BTK mutations associated with resistance to noncovalent BTK inhibitors.Blood Adv. 2023 Oct 10;7(19):5698-5702. doi: 10.1182/bloodadvances.2022008955. Blood Adv. 2023. PMID: 36661329 Free PMC article. No abstract available.
-
HIF-PH Encoded by EGLN1 Is a Potential Therapeutic Target for Chronic Lymphocytic Leukemia.Pharmaceuticals (Basel). 2022 Jun 10;15(6):734. doi: 10.3390/ph15060734. Pharmaceuticals (Basel). 2022. PMID: 35745653 Free PMC article.
-
Pirtobrutinib: a new hope for patients with BTK inhibitor-refractory lymphoproliferative disorders.Blood. 2023 Jun 29;141(26):3137-3142. doi: 10.1182/blood.2023020240. Blood. 2023. PMID: 37156004 Free PMC article.
-
Relapsed/Refractory Chronic Lymphocytic Leukemia (CLL).Curr Hematol Malig Rep. 2023 Oct;18(5):130-143. doi: 10.1007/s11899-023-00700-z. Epub 2023 Jun 6. Curr Hematol Malig Rep. 2023. PMID: 37278884 Free PMC article. Review.
-
Transcriptomic and proteomic differences in BTK-WT and BTK-mutated CLL and their changes during therapy with pirtobrutinib.Blood Adv. 2024 Sep 10;8(17):4487-4501. doi: 10.1182/bloodadvances.2023012360. Blood Adv. 2024. PMID: 38968154 Free PMC article.
References
-
- Mato AR. Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet 2021;397:892–901. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 CA229086/CA/NCI NIH HHS/United States
- T32 CA009207/CA/NCI NIH HHS/United States
- R01 CA228135/CA/NCI NIH HHS/United States
- P50 CA254838/CA/NCI NIH HHS/United States
- F31 CA275378/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 HL159175/HL/NHLBI NIH HHS/United States
- R01 CA242020/CA/NCI NIH HHS/United States
- R01 CA173636/CA/NCI NIH HHS/United States
- K08 CA230319/CA/NCI NIH HHS/United States
- R01 CA216421/CA/NCI NIH HHS/United States
- R01CA216421, R01CA173636, R01CA228135, P01CA229086/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases