

An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials
- PMID: 33926102
- PMCID: PMC8123696
- DOI: 10.3390/ijms22094534
An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials
Abstract
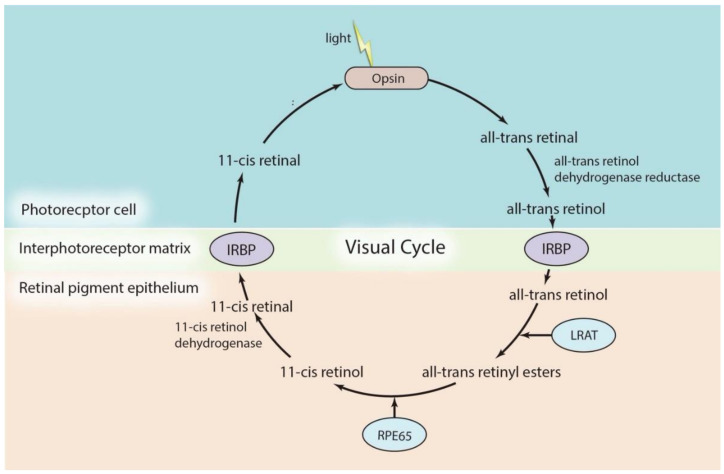
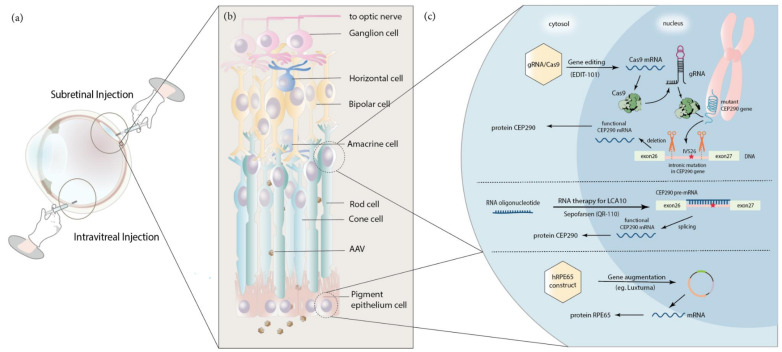
Inherited retinal dystrophies (IRDs) are a group of rare eye diseases caused by gene mutations that result in the degradation of cone and rod photoreceptors or the retinal pigment epithelium. Retinal degradation progress is often irreversible, with clinical manifestations including color or night blindness, peripheral visual defects and subsequent vision loss. Thus, gene therapies that restore functional retinal proteins by either replenishing unmutated genes or truncating mutated genes are needed. Coincidentally, the eye's accessibility and immune-privileged status along with major advances in gene identification and gene delivery systems heralded gene therapies for IRDs. Among these clinical trials, voretigene neparvovec-rzyl (Luxturna), an adeno-associated virus vector-based gene therapy drug, was approved by the FDA for treating patients with confirmed biallelic RPE65 mutation-associated Leber Congenital Amaurosis (LCA) in 2017. This review includes current IRD gene therapy clinical trials and further summarizes preclinical studies and therapeutic strategies for LCA, including adeno-associated virus-based gene augmentation therapy, 11-cis-retinal replacement, RNA-based antisense oligonucleotide therapy and CRISPR-Cas9 gene-editing therapy. Understanding the gene therapy development for LCA may accelerate and predict the potential hurdles of future therapeutics translation. It may also serve as the template for the research and development of treatment for other IRDs.
Keywords: Leber Congenital Amaurosis; RNA-based antisense oligonucleotide therapy; gene augmentation therapy; gene-editing therapy; inherited retinal dystrophy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Gene Therapy in Hereditary Retinal Dystrophies: The Usefulness of Diagnostic Tools in Candidate Patient Selections.Int J Mol Sci. 2023 Sep 6;24(18):13756. doi: 10.3390/ijms241813756. Int J Mol Sci. 2023. PMID: 37762059 Free PMC article. Review.
-
Gene therapy for inherited retinal diseases: progress and possibilities.Clin Exp Optom. 2021 May;104(4):444-454. doi: 10.1080/08164622.2021.1880863. Epub 2021 Mar 2. Clin Exp Optom. 2021. PMID: 33689657 Review.
-
RNA-Based Therapeutic Strategies for Inherited Retinal Dystrophies.Adv Exp Med Biol. 2019;1185:71-77. doi: 10.1007/978-3-030-27378-1_12. Adv Exp Med Biol. 2019. PMID: 31884591 Review.
-
Gene therapy for Leber congenital amaurosis: advances and future directions.Graefes Arch Clin Exp Ophthalmol. 2012 Aug;250(8):1117-28. doi: 10.1007/s00417-012-2028-2. Epub 2012 May 29. Graefes Arch Clin Exp Ophthalmol. 2012. PMID: 22644094 Free PMC article. Review.
-
Gene therapy for RPE65-related retinal disease.Ophthalmic Genet. 2018 Dec;39(6):671-677. doi: 10.1080/13816810.2018.1533027. Epub 2018 Oct 18. Ophthalmic Genet. 2018. PMID: 30335549 Review.
Cited by
-
Towards an Understanding of Retinal Diseases and Novel Treatment.Int J Mol Sci. 2022 Jul 8;23(14):7576. doi: 10.3390/ijms23147576. Int J Mol Sci. 2022. PMID: 35886925 Free PMC article.
-
A multidisciplinary approach to inherited retinal dystrophies from diagnosis to initial care: a narrative review with inputs from clinical practice.Orphanet J Rare Dis. 2023 Jul 31;18(1):223. doi: 10.1186/s13023-023-02798-z. Orphanet J Rare Dis. 2023. PMID: 37525225 Free PMC article. Review.
-
Navigating the Genetic Landscape: A Comprehensive Review of Novel Therapeutic Strategies for Retinitis Pigmentosa Management.Cureus. 2024 Aug 16;16(8):e67046. doi: 10.7759/cureus.67046. eCollection 2024 Aug. Cureus. 2024. PMID: 39286723 Free PMC article. Review.
-
Compact Cas9d and HEARO enzymes for genome editing discovered from uncultivated microbes.Nat Commun. 2022 Dec 15;13(1):7602. doi: 10.1038/s41467-022-35257-7. Nat Commun. 2022. PMID: 36522342 Free PMC article.
-
Structural Differences Across Multiple Visual Cortical Regions in the Absence of Cone Function in Congenital Achromatopsia.Front Neurosci. 2021 Oct 14;15:718958. doi: 10.3389/fnins.2021.718958. eCollection 2021. Front Neurosci. 2021. PMID: 34720857 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
