

Evidence for 28 genetic disorders discovered by combining healthcare and research data
- PMID: 33057194
- PMCID: PMC7116826
- DOI: 10.1038/s41586-020-2832-5
Evidence for 28 genetic disorders discovered by combining healthcare and research data
Abstract
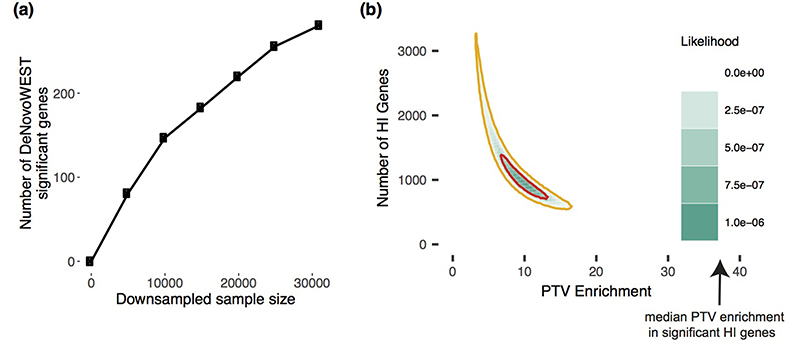
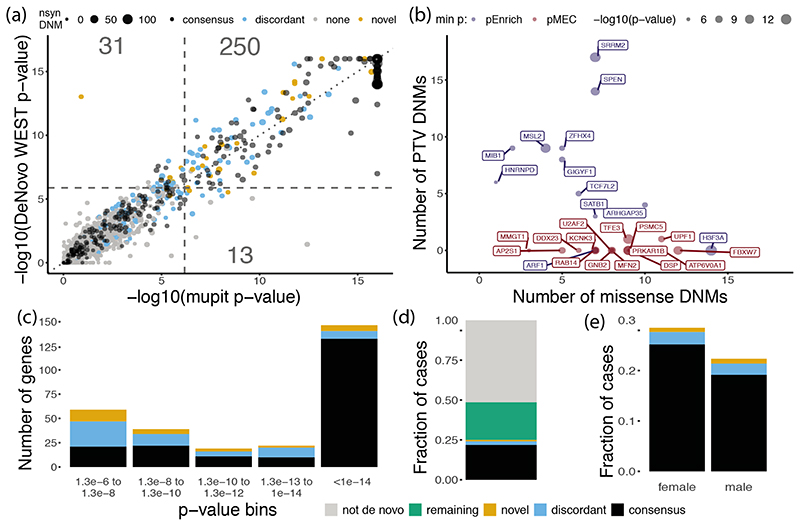
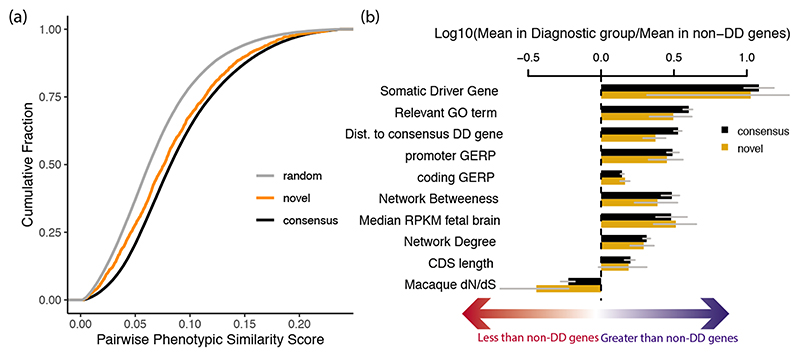
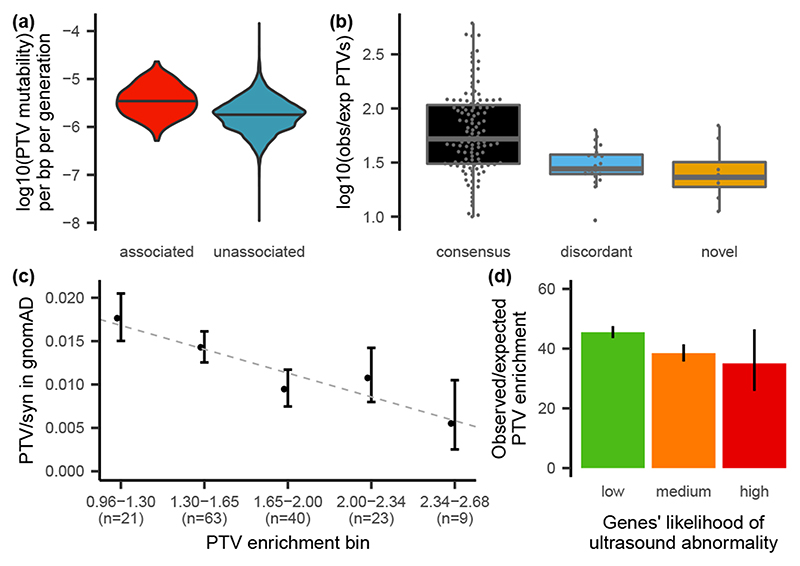
De novo mutations in protein-coding genes are a well-established cause of developmental disorders1. However, genes known to be associated with developmental disorders account for only a minority of the observed excess of such de novo mutations1,2. Here, to identify previously undescribed genes associated with developmental disorders, we integrate healthcare and research exome-sequence data from 31,058 parent-offspring trios of individuals with developmental disorders, and develop a simulation-based statistical test to identify gene-specific enrichment of de novo mutations. We identified 285 genes that were significantly associated with developmental disorders, including 28 that had not previously been robustly associated with developmental disorders. Although we detected more genes associated with developmental disorders, much of the excess of de novo mutations in protein-coding genes remains unaccounted for. Modelling suggests that more than 1,000 genes associated with developmental disorders have not yet been described, many of which are likely to be less penetrant than the currently known genes. Research access to clinical diagnostic datasets will be critical for completing the map of genes associated with developmental disorders.
Conflict of interest statement
Z.Z., K.J.A., R.I.T., J.J., and K.R. are employees of GeneDx. J.J. and K.R. are shareholders of OPKO. M.E.H. is a co-founder of, consultant to, and holds shares in, Congenica Ltd, a genetics diagnostic company.
Figures
Similar articles
-
A Mild PUM1 Mutation Is Associated with Adult-Onset Ataxia, whereas Haploinsufficiency Causes Developmental Delay and Seizures.Cell. 2018 Feb 22;172(5):924-936.e11. doi: 10.1016/j.cell.2018.02.006. Cell. 2018. PMID: 29474920 Free PMC article.
-
Exome-wide assessment of the functional impact and pathogenicity of multinucleotide mutations.Genome Res. 2019 Jul;29(7):1047-1056. doi: 10.1101/gr.239756.118. Epub 2019 Jun 21. Genome Res. 2019. PMID: 31227601 Free PMC article.
-
Penetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders.Eur J Med Genet. 2015 Dec;58(12):715-8. doi: 10.1016/j.ejmg.2015.10.007. Epub 2015 Oct 24. Eur J Med Genet. 2015. PMID: 26506440
-
DECIPHER: web-based, community resource for clinical interpretation of rare variants in developmental disorders.Hum Mol Genet. 2012 Oct 15;21(R1):R37-44. doi: 10.1093/hmg/dds362. Epub 2012 Sep 8. Hum Mol Genet. 2012. PMID: 22962312 Free PMC article. Review.
-
Genomically Aided Diagnosis of Severe Developmental Disorders.Annu Rev Genomics Hum Genet. 2020 Aug 31;21:327-349. doi: 10.1146/annurev-genom-120919-082329. Epub 2020 May 18. Annu Rev Genomics Hum Genet. 2020. PMID: 32421356 Review.
Cited by
-
Federated analysis of autosomal recessive coding variants in 29,745 developmental disorder patients from diverse populations.Nat Genet. 2024 Oct;56(10):2046-2053. doi: 10.1038/s41588-024-01910-8. Epub 2024 Sep 23. Nat Genet. 2024. PMID: 39313616 Free PMC article.
-
The genetic cause of neurodevelopmental disorders in 30 consanguineous families.Front Med (Lausanne). 2024 Aug 30;11:1424753. doi: 10.3389/fmed.2024.1424753. eCollection 2024. Front Med (Lausanne). 2024. PMID: 39281811 Free PMC article.
-
Epigenomic dysregulation correlates with arachnoid cyst formation and neurodevelopmental symptoms.Nat Med. 2023 Mar;29(3):541-542. doi: 10.1038/s41591-023-02239-1. Nat Med. 2023. PMID: 36932244 Free PMC article.
-
The frequency of somatic mutations in cancer predicts the phenotypic relevance of germline mutations.Front Genet. 2023 Jan 9;13:1045301. doi: 10.3389/fgene.2022.1045301. eCollection 2022. Front Genet. 2023. PMID: 36699457 Free PMC article.
-
Variability in Phelan-McDermid Syndrome in a Cohort of 210 Individuals.Front Genet. 2022 Apr 12;13:652454. doi: 10.3389/fgene.2022.652454. eCollection 2022. Front Genet. 2022. PMID: 35495150 Free PMC article.
References
-
- Samocha KE, et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv. 2017 doi: 10.1101/148353. 148353. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
