

MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis
- PMID: 31834374
- PMCID: PMC7962909
- DOI: 10.1093/brain/awz379
MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis
Erratum in
-
Erratum.Brain. 2020 Mar 1;143(3):e24. doi: 10.1093/brain/awaa007. Brain. 2020. PMID: 32333675 Free PMC article. No abstract available.
Abstract
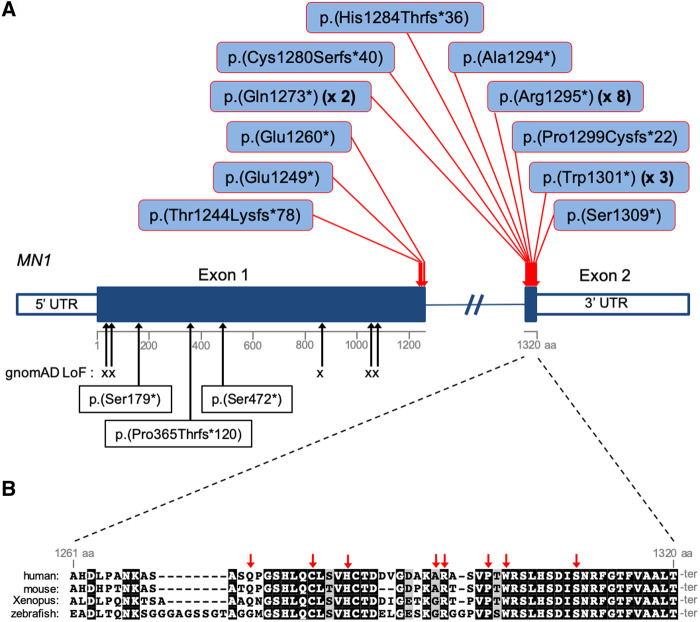
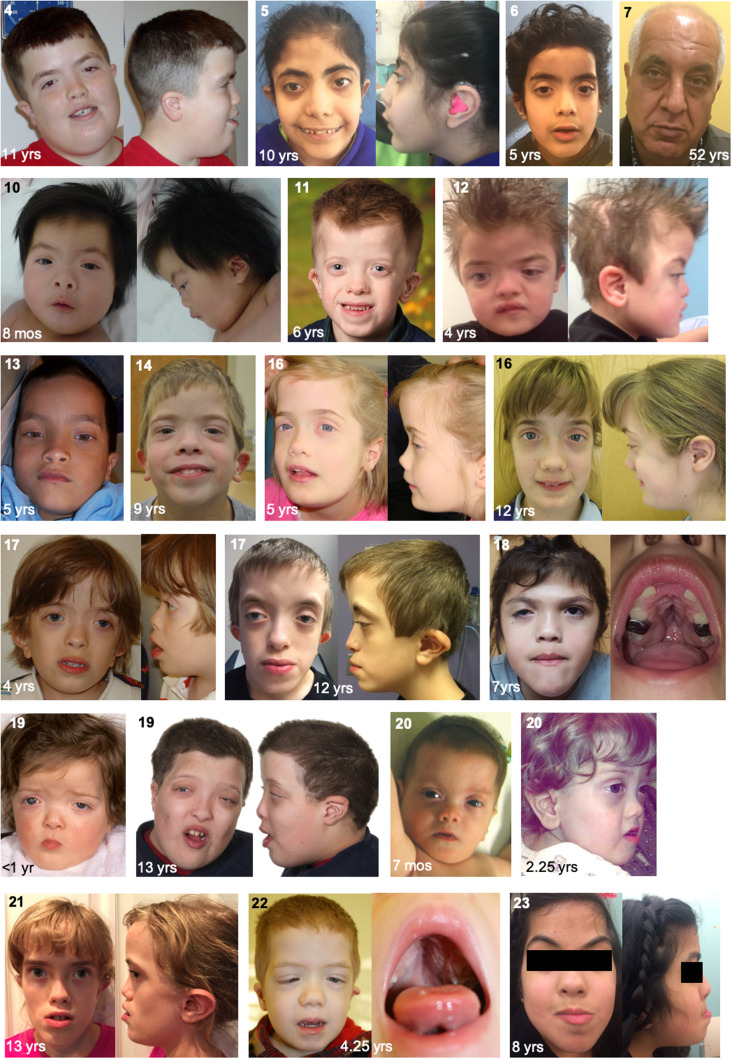
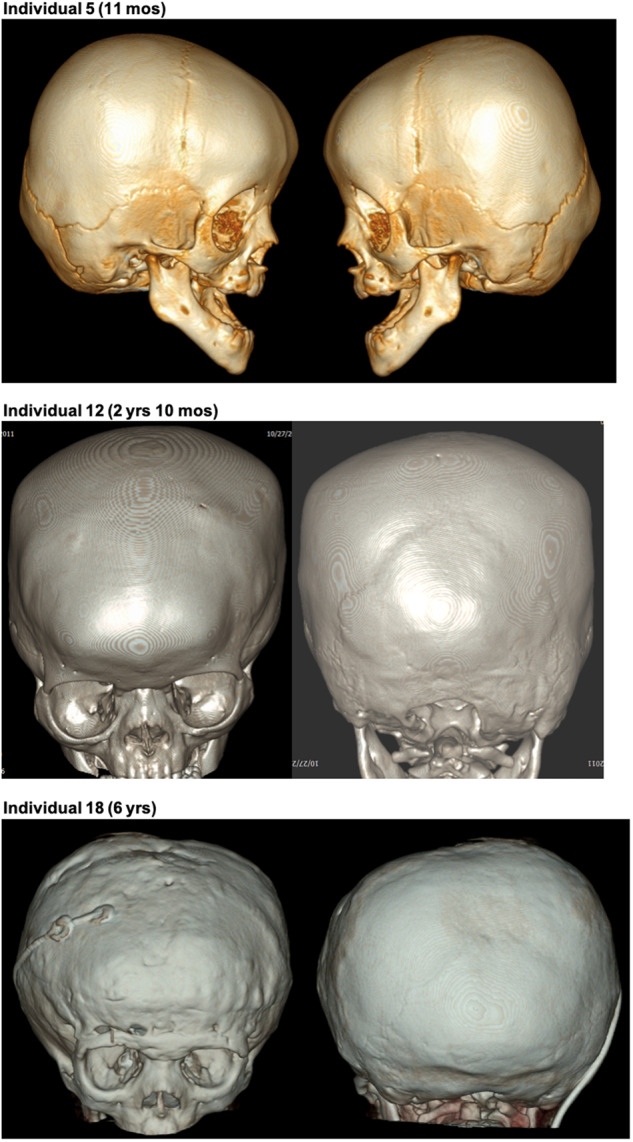
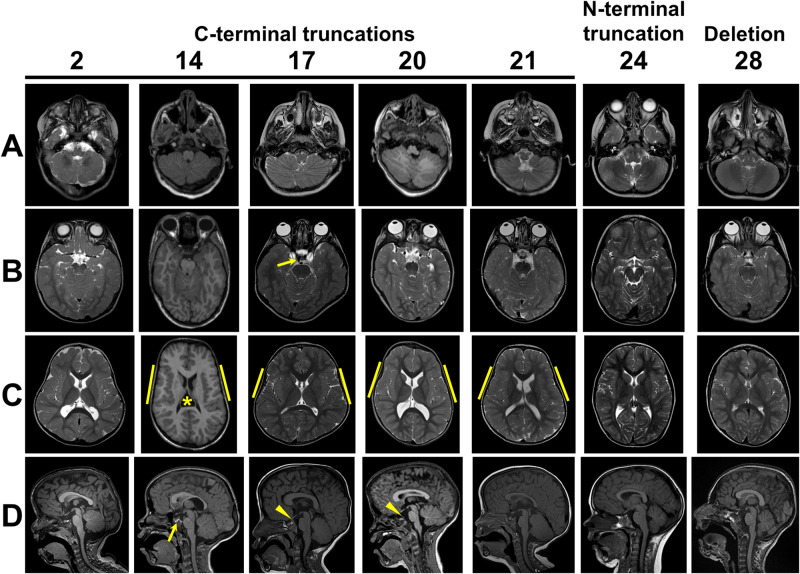
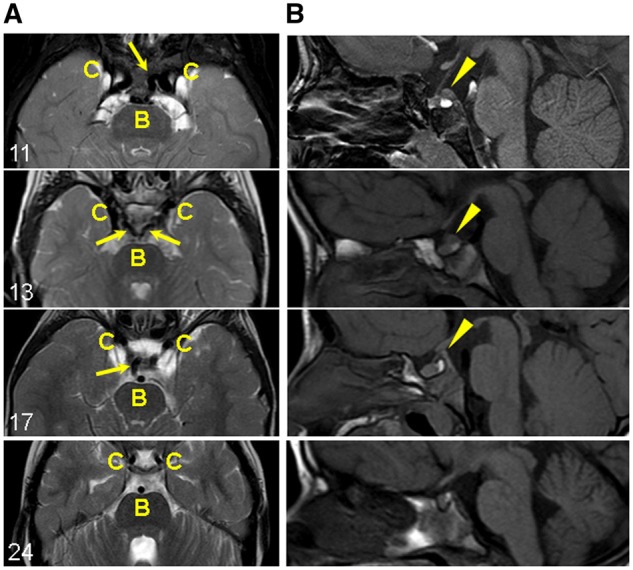
MN1 encodes a transcriptional co-regulator without homology to other proteins, previously implicated in acute myeloid leukaemia and development of the palate. Large deletions encompassing MN1 have been reported in individuals with variable neurodevelopmental anomalies and non-specific facial features. We identified a cluster of de novo truncating mutations in MN1 in a cohort of 23 individuals with strikingly similar dysmorphic facial features, especially midface hypoplasia, and intellectual disability with severe expressive language delay. Imaging revealed an atypical form of rhombencephalosynapsis, a distinctive brain malformation characterized by partial or complete loss of the cerebellar vermis with fusion of the cerebellar hemispheres, in 8/10 individuals. Rhombencephalosynapsis has no previously known definitive genetic or environmental causes. Other frequent features included perisylvian polymicrogyria, abnormal posterior clinoid processes and persistent trigeminal artery. MN1 is encoded by only two exons. All mutations, including the recurrent variant p.Arg1295* observed in 8/21 probands, fall in the terminal exon or the extreme 3' region of exon 1, and are therefore predicted to result in escape from nonsense-mediated mRNA decay. This was confirmed in fibroblasts from three individuals. We propose that the condition described here, MN1 C-terminal truncation (MCTT) syndrome, is not due to MN1 haploinsufficiency but rather is the result of dominantly acting C-terminally truncated MN1 protein. Our data show that MN1 plays a critical role in human craniofacial and brain development, and opens the door to understanding the biological mechanisms underlying rhombencephalosynapsis.
Keywords: MCTT syndrome; MN1; craniofacial development; intellectual disability; rhombencephalosynapsis.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures

Comment in
-
MN1 gene loss-of-function mutation causes cleft palate in a pedigree.Brain. 2021 Mar 3;144(2):e18. doi: 10.1093/brain/awaa431. Brain. 2021. PMID: 33351070 Free PMC article. No abstract available.
-
Reply: MN1 gene loss-of-function mutation causes cleft palate in a pedigree.Brain. 2021 Mar 3;144(2):e19. doi: 10.1093/brain/awaa432. Brain. 2021. PMID: 33351141 No abstract available.
Similar articles
-
A Novel Pathogenic Variant in the MN1 Gene in a Patient Presenting with Rhombencephalosynapsis and Craniofacial Anomalies, Expanding MN1 C-terminal Truncation Syndrome.J Pediatr Genet. 2021 Apr 14;12(3):254-257. doi: 10.1055/s-0041-1728650. eCollection 2023 Sep. J Pediatr Genet. 2021. PMID: 37575653 Free PMC article.
-
Novel truncating variant of MN1 penultimate exon identified in a Chinese patient with newly recognized MN1 C-terminal truncation syndrome: Case report and literature review.Int J Dev Neurosci. 2022 Feb;82(1):96-103. doi: 10.1002/jdn.10154. Epub 2021 Nov 4. Int J Dev Neurosci. 2022. PMID: 34708882
-
Prenatal diagnosis of rhombencephalosynapsis: neuroimaging features and severity of vermian anomaly.Ultrasound Obstet Gynecol. 2021 Dec;58(6):864-874. doi: 10.1002/uog.23660. Ultrasound Obstet Gynecol. 2021. PMID: 33942916
-
Distinctive Craniofacial and Oral Anomalies in MN1 C-terminal Truncation Syndrome.Chin J Dent Res. 2024 Mar 28;27(1):47-52. doi: 10.3290/j.cjdr.b5128655. Chin J Dent Res. 2024. PMID: 38546519 Review.
-
Microdeletion del(22)(q12.2) encompassing the facial development-associated gene, MN1 (meningioma 1) in a child with Pierre-Robin sequence (including cleft palate) and neurofibromatosis 2 (NF2): a case report and review of the literature.BMC Med Genet. 2012 Mar 22;13:19. doi: 10.1186/1471-2350-13-19. BMC Med Genet. 2012. PMID: 22436304 Free PMC article. Review.
Cited by
-
MN1 overexpression with varying tumor grade is a promising predictor of survival of glioma patients.Hum Mol Genet. 2021 Jan 6;29(21):3532-3545. doi: 10.1093/hmg/ddaa231. Hum Mol Genet. 2021. PMID: 33105486 Free PMC article.
-
Systematic analysis of variants escaping nonsense-mediated decay uncovers candidate Mendelian diseases.Am J Hum Genet. 2024 Jan 4;111(1):70-81. doi: 10.1016/j.ajhg.2023.11.007. Epub 2023 Dec 12. Am J Hum Genet. 2024. PMID: 38091987 Free PMC article.
-
MN1 gene loss-of-function mutation causes cleft palate in a pedigree.Brain. 2021 Mar 3;144(2):e18. doi: 10.1093/brain/awaa431. Brain. 2021. PMID: 33351070 Free PMC article. No abstract available.
-
Combining globally search for a regular expression and print matching lines with bibliographic monitoring of genomic database improves diagnosis.Front Genet. 2023 Apr 20;14:1122985. doi: 10.3389/fgene.2023.1122985. eCollection 2023. Front Genet. 2023. PMID: 37152996 Free PMC article.
-
Editorial: Sub-molecular mechanism of genetic epilepsy.Front Mol Neurosci. 2022 Jul 26;15:958747. doi: 10.3389/fnmol.2022.958747. eCollection 2022. Front Mol Neurosci. 2022. PMID: 35959103 Free PMC article. No abstract available.
References
-
- Beck M, Peterson JF, McConnell J, McGuire M, Asato M, Losee JE, et al.Craniofacial abnormalities and developmental delay in two families with overlapping 22q12.1 microdeletions involving the MN1 gene. Am J Med Genet A 2015; 167A: 1047–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
