

Statistical investigations of protein residue direct couplings
- PMID: 30596639
- PMCID: PMC6329532
- DOI: 10.1371/journal.pcbi.1006237
Statistical investigations of protein residue direct couplings
Abstract
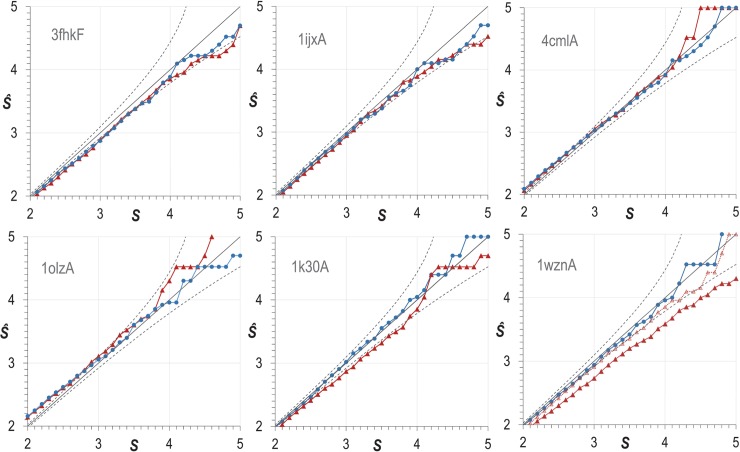
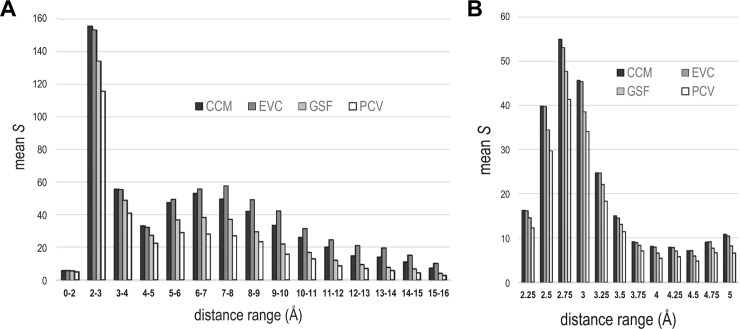
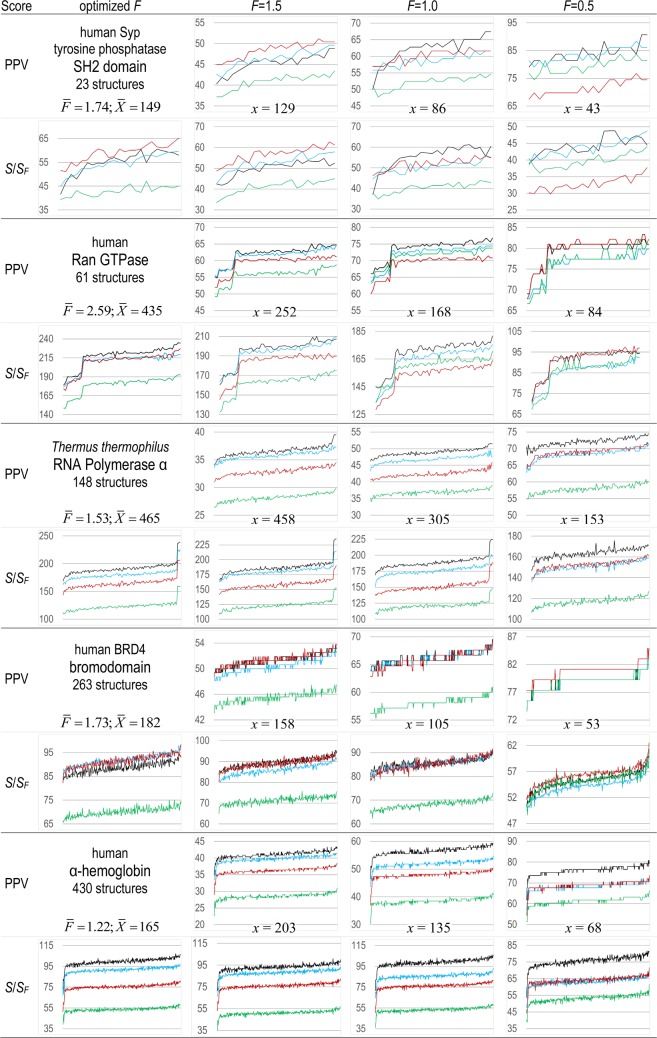
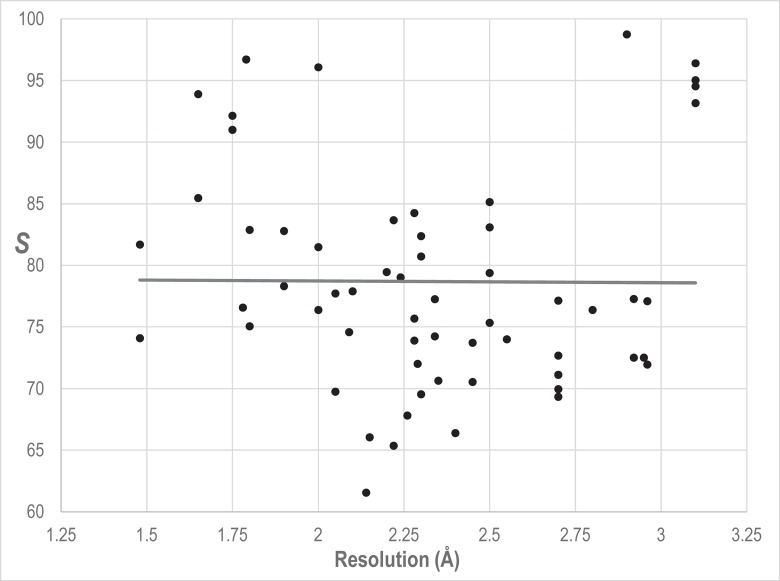
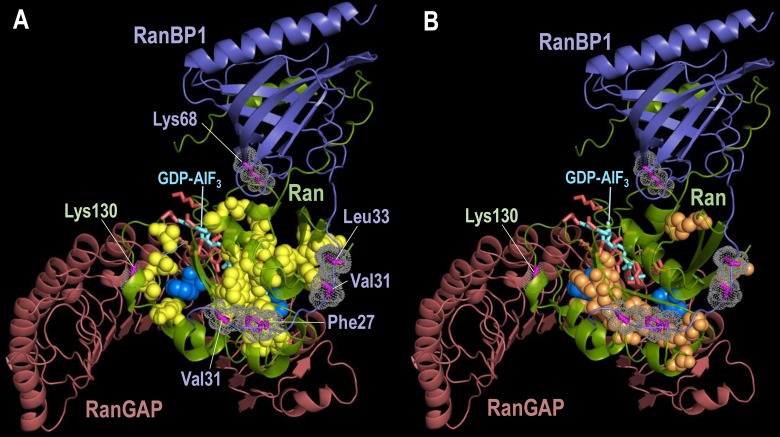
Protein Direct Coupling Analysis (DCA), which predicts residue-residue contacts based on covarying positions within a multiple sequence alignment, has been remarkably effective. This suggests that there is more to learn from sequence correlations than is generally assumed, and calls for deeper investigations into DCA and perhaps into other types of correlations. Here we describe an approach that enables such investigations by measuring, as an estimated p-value, the statistical significance of the association between residue-residue covariance and structural interactions, either internal or homodimeric. Its application to thirty protein superfamilies confirms that direct coupling (DC) scores correlate with 3D pairwise contacts with very high significance. This method also permits quantitative assessment of the relative performance of alternative DCA methods, and of the degree to which they detect direct versus indirect couplings. We illustrate its use to assess, for a given protein, the biological relevance of alternative conformational states, to investigate the possible mechanistic implications of differences between these states, and to characterize subtle aspects of direct couplings. Our analysis indicates that direct pairwise correlations may be largely distinct from correlated patterns associated with functional specialization, and that the joint analysis of both types of correlations can yield greater power. Data, programs, and source code are freely available at http://evaldca.igs.umaryland.edu.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Adaptive Smith-Waterman residue match seeding for protein structural alignment.Proteins. 2013 Oct;81(10):1823-39. doi: 10.1002/prot.24327. Epub 2013 Aug 19. Proteins. 2013. PMID: 23720362
-
Direct coupling analysis for protein contact prediction.Methods Mol Biol. 2014;1137:55-70. doi: 10.1007/978-1-4939-0366-5_5. Methods Mol Biol. 2014. PMID: 24573474
-
Covariation analysis of local amino acid sequences in recurrent protein local structures.J Bioinform Comput Biol. 2005 Dec;3(6):1391-409. doi: 10.1142/s0219720005001648. J Bioinform Comput Biol. 2005. PMID: 16374913
-
Five hierarchical levels of sequence-structure correlation in proteins.Appl Bioinformatics. 2004;3(2-3):97-104. doi: 10.2165/00822942-200403020-00004. Appl Bioinformatics. 2004. PMID: 15693735 Review.
-
Gleaning structural and functional information from correlations in protein multiple sequence alignments.Curr Opin Struct Biol. 2016 Jun;38:1-8. doi: 10.1016/j.sbi.2016.04.006. Epub 2016 May 12. Curr Opin Struct Biol. 2016. PMID: 27179293 Review.
Cited by
-
SPARC: Structural properties associated with residue constraints.Comput Struct Biotechnol J. 2022 Apr 7;20:1702-1715. doi: 10.1016/j.csbj.2022.04.005. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35495120 Free PMC article.
-
Obtaining extremely large and accurate protein multiple sequence alignments from curated hierarchical alignments.Database (Oxford). 2020 Jan 1;2020:baaa042. doi: 10.1093/database/baaa042. Database (Oxford). 2020. PMID: 32500917 Free PMC article.
-
Episodic evolution of coadapted sets of amino acid sites in mitochondrial proteins.PLoS Genet. 2021 Jan 25;17(1):e1008711. doi: 10.1371/journal.pgen.1008711. eCollection 2021 Jan. PLoS Genet. 2021. PMID: 33493156 Free PMC article.
-
Structural insights into the elevator-type transport mechanism of a bacterial ZIP metal transporter.Nat Commun. 2023 Jan 24;14(1):385. doi: 10.1038/s41467-023-36048-4. Nat Commun. 2023. PMID: 36693843 Free PMC article.
-
Deep Analysis of Residue Constraints (DARC): identifying determinants of protein functional specificity.Sci Rep. 2020 Feb 3;10(1):1691. doi: 10.1038/s41598-019-55118-6. Sci Rep. 2020. PMID: 32015389 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
