

MicroScope-an integrated resource for community expertise of gene functions and comparative analysis of microbial genomic and metabolic data
- PMID: 28968784
- PMCID: PMC6931091
- DOI: 10.1093/bib/bbx113
MicroScope-an integrated resource for community expertise of gene functions and comparative analysis of microbial genomic and metabolic data
Abstract
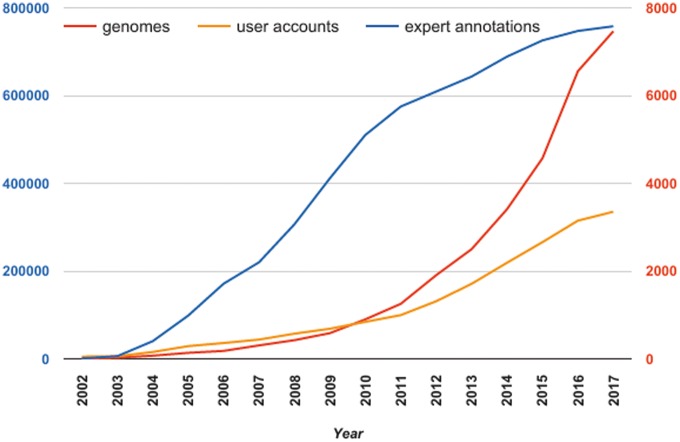
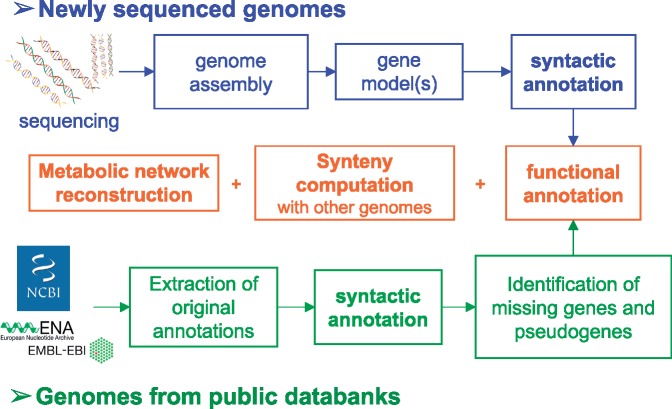
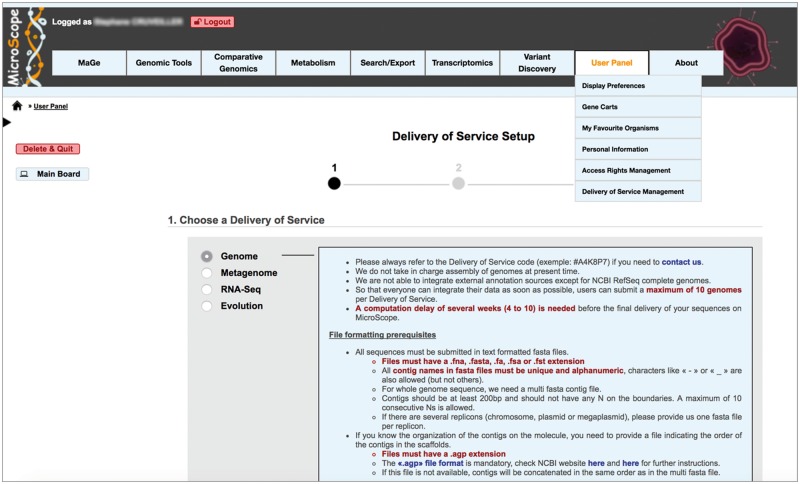
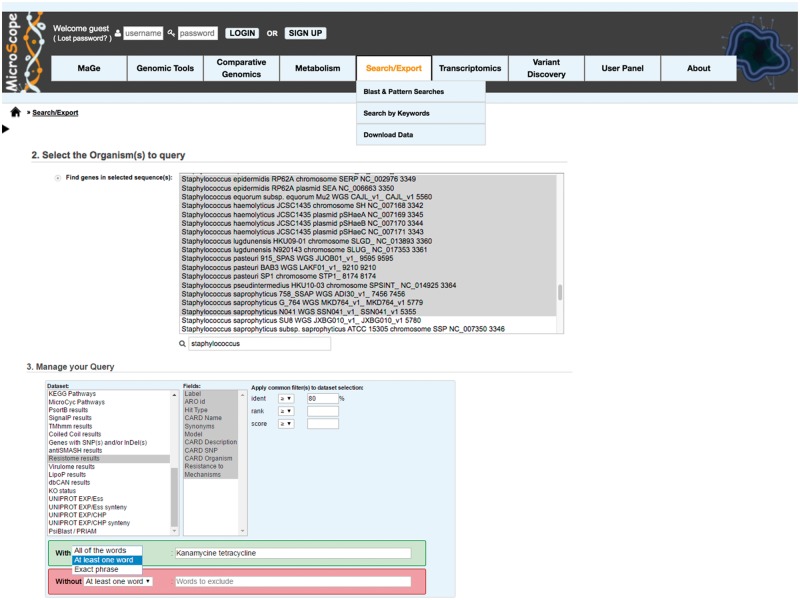
The overwhelming list of new bacterial genomes becoming available on a daily basis makes accurate genome annotation an essential step that ultimately determines the relevance of thousands of genomes stored in public databanks. The MicroScope platform (http://www.genoscope.cns.fr/agc/microscope) is an integrative resource that supports systematic and efficient revision of microbial genome annotation, data management and comparative analysis. Starting from the results of our syntactic, functional and relational annotation pipelines, MicroScope provides an integrated environment for the expert annotation and comparative analysis of prokaryotic genomes. It combines tools and graphical interfaces to analyze genomes and to perform the manual curation of gene function in a comparative genomics and metabolic context. In this article, we describe the free-of-charge MicroScope services for the annotation and analysis of microbial (meta)genomes, transcriptomic and re-sequencing data. Then, the functionalities of the platform are presented in a way providing practical guidance and help to the nonspecialists in bioinformatics. Newly integrated analysis tools (i.e. prediction of virulence and resistance genes in bacterial genomes) and original method recently developed (the pan-genome graph representation) are also described. Integrated environments such as MicroScope clearly contribute, through the user community, to help maintaining accurate resources.
Keywords: comparative genomics; gene function curation; metabolic networks; microbial genome annotation system; transcriptomics; variant detection.
© The Author 2017. Published by Oxford University Press.
Figures
Similar articles
-
MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes.Nucleic Acids Res. 2017 Jan 4;45(D1):D517-D528. doi: 10.1093/nar/gkw1101. Epub 2016 Nov 29. Nucleic Acids Res. 2017. PMID: 27899624 Free PMC article.
-
MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis.Nucleic Acids Res. 2020 Jan 8;48(D1):D579-D589. doi: 10.1093/nar/gkz926. Nucleic Acids Res. 2020. PMID: 31647104 Free PMC article.
-
MicroScope--an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data.Nucleic Acids Res. 2013 Jan;41(Database issue):D636-47. doi: 10.1093/nar/gks1194. Epub 2012 Nov 27. Nucleic Acids Res. 2013. PMID: 23193269 Free PMC article.
-
An Experimental Approach to Genome Annotation: This report is based on a colloquium sponsored by the American Academy of Microbiology held July 19-20, 2004, in Washington, DC.Washington (DC): American Society for Microbiology; 2004. Washington (DC): American Society for Microbiology; 2004. PMID: 33001599 Free Books & Documents. Review.
-
Mitochondrial Disease Sequence Data Resource (MSeqDR): a global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities.Mol Genet Metab. 2015 Mar;114(3):388-96. doi: 10.1016/j.ymgme.2014.11.016. Epub 2014 Dec 4. Mol Genet Metab. 2015. PMID: 25542617 Free PMC article. Review.
Cited by
-
Artificial intelligence-based prediction of pathogen emergence and evolution in the world of synthetic biology.Microb Biotechnol. 2024 Oct;17(10):e70014. doi: 10.1111/1751-7915.70014. Microb Biotechnol. 2024. PMID: 39364593 Free PMC article. Review.
-
Insights into the Diversity of Secondary Metabolites of Planktothrix Using a Biphasic Approach Combining Global Genomics and Metabolomics.Toxins (Basel). 2019 Aug 27;11(9):498. doi: 10.3390/toxins11090498. Toxins (Basel). 2019. PMID: 31461939 Free PMC article.
-
Ammonia Oxidation by the Arctic Terrestrial Thaumarchaeote Candidatus Nitrosocosmicus arcticus Is Stimulated by Increasing Temperatures.Front Microbiol. 2019 Jul 17;10:1571. doi: 10.3389/fmicb.2019.01571. eCollection 2019. Front Microbiol. 2019. PMID: 31379764 Free PMC article.
-
Genetic diversity and population structure of Tenacibaculum maritimum, a serious bacterial pathogen of marine fish: from genome comparisons to high throughput MALDI-TOF typing.Vet Res. 2020 May 7;51(1):60. doi: 10.1186/s13567-020-00782-0. Vet Res. 2020. PMID: 32381115 Free PMC article.
-
Channeling C1 Metabolism toward S-Adenosylmethionine-Dependent Conversion of Estrogens to Androgens in Estrogen-Degrading Bacteria.mBio. 2020 Aug 25;11(4):e01259-20. doi: 10.1128/mBio.01259-20. mBio. 2020. PMID: 32843544 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
