

Survival in infants treated with sebelipase Alfa for lysosomal acid lipase deficiency: an open-label, multicenter, dose-escalation study
- PMID: 28179030
- PMCID: PMC5299659
- DOI: 10.1186/s13023-017-0587-3
Survival in infants treated with sebelipase Alfa for lysosomal acid lipase deficiency: an open-label, multicenter, dose-escalation study
Abstract
Background: Infants presenting with lysosomal acid lipase deficiency have marked failure to thrive, diarrhea, massive hepatosplenomegaly, anemia, rapidly progressive liver disease, and death typically in the first 6 months of life; the only available potential treatment has been hematopoietic stem cell transplantation, which is associated with high morbidity and mortality in this population. The study objective was to evaluate safety and efficacy (including survival) of enzyme replacement with sebelipase alfa in infants with lysosomal acid lipase deficiency. This is an ongoing multicenter, open-label, phase 2/3 study conducted in nine countries. The study enrolled infants with growth failure prior to 6 months of age with rapidly progressive lysosomal acid lipase deficiency; they received once-weekly doses of sebelipase alfa initiated at 0.35 mg/kg with intrapatient dose escalation up to 5 mg/kg. The main outcome of interest is survival to 12 months and survival beyond 24 months of age.
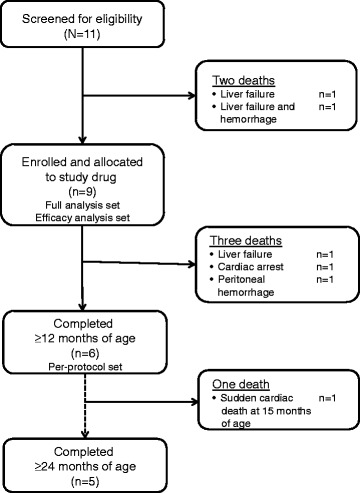
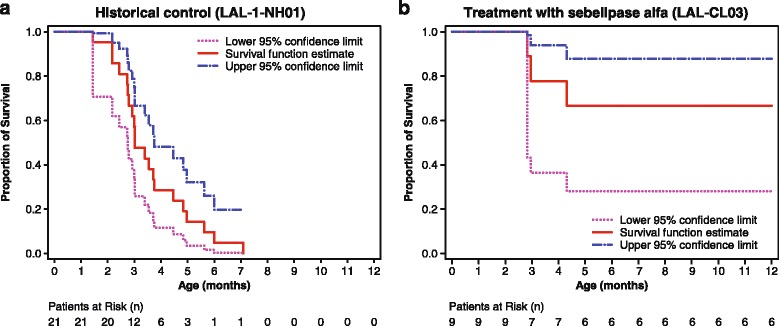
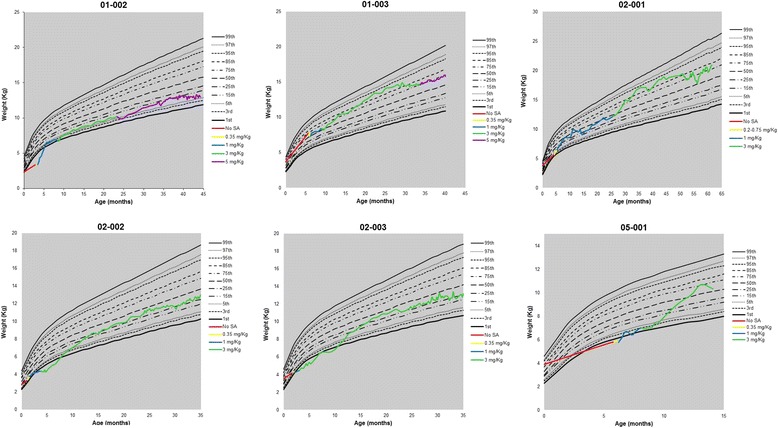
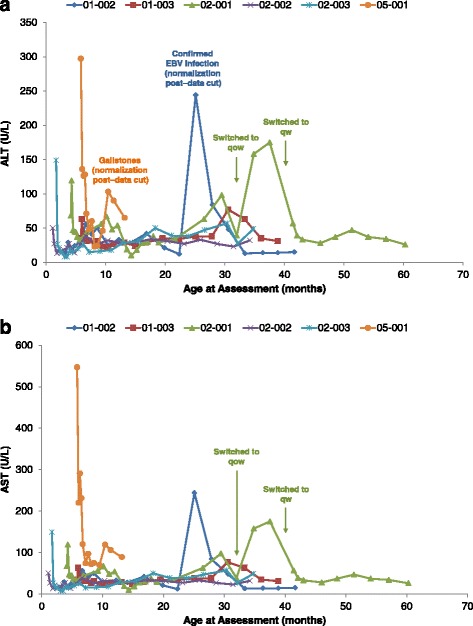
Results: Nine patients were enrolled; median age at baseline was 3.0 months (range 1.1-5.8 months). Sixty-seven percent (exact 95% CI 30%-93%) of sebelipase alfa-treated infants survived to 12 months of age compared with 0% (exact 95% CI 0%-16%) for a historical control group (n = 21). Patients who survived to age 12 months exhibited improvements in weight-for-age, reductions in markers of liver dysfunction and hepatosplenomegaly, and improvements in anemia and gastrointestinal symptoms. Three deaths occurred early (first few months of life), two patients died because of advanced disease, and a third patient died following complications of non-protocol-specified abdominal paracentesis. A fourth death occurred at 15 months of age and was related to other clinical conditions. The five surviving patients have survived to age ≥24 months with continued sebelipase alfa treatment; all have displayed marked improvement in growth parameters and liver function. Serious adverse events considered related to sebelipase alfa were reported in one of the nine infants (infusion reaction: tachycardia, pallor, chills, and pyrexia). Most infusion-associated reactions were mild and non-serious.
Conclusion: Sebelipase alfa markedly improved survival with substantial clinically meaningful improvements in growth and other key disease manifestations in infants with rapidly progressive lysosomal acid lipase deficiency TRIAL REGISTRATION: Clinicaltrials.gov NCT01371825 . Registered 9 June 2011.
Keywords: Hemophagocytic lymphohistiocytosis; Lysosomal acid lipase deficiency; Sebelipase alfa; Survival; Wolman disease.
Figures
Similar articles
-
Long-term survival with sebelipase alfa enzyme replacement therapy in infants with rapidly progressive lysosomal acid lipase deficiency: final results from 2 open-label studies.Orphanet J Rare Dis. 2021 Jan 6;16(1):13. doi: 10.1186/s13023-020-01577-4. Orphanet J Rare Dis. 2021. PMID: 33407676 Free PMC article.
-
Sebelipase Alfa: A Review in Lysosomal Acid Lipase Deficiency.Am J Cardiovasc Drugs. 2016 Dec;16(6):461-468. doi: 10.1007/s40256-016-0203-2. Am J Cardiovasc Drugs. 2016. PMID: 27878737 Review.
-
Early onset lysosomal acid lipase deficiency presenting as secondary hemophagocytic lymphohistiocytosis: Two infants treated with sebelipase alfa.Clin Res Hepatol Gastroenterol. 2018 Oct;42(5):e77-e82. doi: 10.1016/j.clinre.2018.03.012. Epub 2018 Apr 26. Clin Res Hepatol Gastroenterol. 2018. PMID: 29705274
-
Sebelipase alfa for lysosomal acid lipase deficiency: 5-year treatment experience from a phase 2 open-label extension study.Liver Int. 2020 Sep;40(9):2203-2214. doi: 10.1111/liv.14603. Epub 2020 Aug 9. Liver Int. 2020. PMID: 32657505 Free PMC article. Clinical Trial.
-
Sebelipase alfa: enzymatic replacement treatment for lysosomal acid lipase deficiency.Drugs Today (Barc). 2016 May;52(5):287-93. doi: 10.1358/dot.2016.52.5.2488974. Drugs Today (Barc). 2016. PMID: 27376161 Review.
Cited by
-
Specific Substrate for the Assay of Lysosomal Acid Lipase.Clin Chem. 2018 Apr;64(4):690-696. doi: 10.1373/clinchem.2017.282251. Epub 2018 Jan 16. Clin Chem. 2018. PMID: 29339442 Free PMC article.
-
Unravelling new pathways of sterol metabolism: lessons learned from in-born errors and cancer.Curr Opin Clin Nutr Metab Care. 2018 Mar;21(2):90-96. doi: 10.1097/MCO.0000000000000442. Curr Opin Clin Nutr Metab Care. 2018. PMID: 29227331 Free PMC article. Review.
-
Recommendations on clinical trial design for treatment of Mucopolysaccharidosis Type III.Orphanet J Rare Dis. 2017 Jun 26;12(1):117. doi: 10.1186/s13023-017-0675-4. Orphanet J Rare Dis. 2017. PMID: 28651568 Free PMC article.
-
Persistent dyslipidemia in treatment of lysosomal acid lipase deficiency.Orphanet J Rare Dis. 2020 Feb 24;15(1):58. doi: 10.1186/s13023-020-1328-6. Orphanet J Rare Dis. 2020. PMID: 32093730 Free PMC article.
-
LC-MS/MS-based enzyme assay for lysosomal acid lipase using dried blood spots.Mol Genet Metab Rep. 2022 Aug 26;33:100913. doi: 10.1016/j.ymgmr.2022.100913. eCollection 2022 Dec. Mol Genet Metab Rep. 2022. PMID: 36065451 Free PMC article.
References
-
- Lysosomal acid lipase deficiency (#278000). Online Mendelian Inheritance in Man web site: omim.org. Accessed 19 Sept 2016.
-
- Grabowski GA, Charnas L, Du H. Lysosomal acid lipase deficiencies: the Wolman disease/cholesteryl ester storage disease spectrum. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, editors. Metabolic and molecular bases of inherited disease. 8. New York, NY: McGraw-Hill; 2012.
-
- Hoffman EP, Barr ML, Giovanni MA, Murray MF, et al. Lysosomal acid lipase deficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993.
-
- Abramov A, Schorr S, Wolman M. Generalized xanthomatosis with calcified adrenals. AMA J Dis Child. 1956;91:282–286. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
