

Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome
- PMID: 28165343
- PMCID: PMC5330744
- DOI: 10.1172/JCI90171
Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome
Abstract
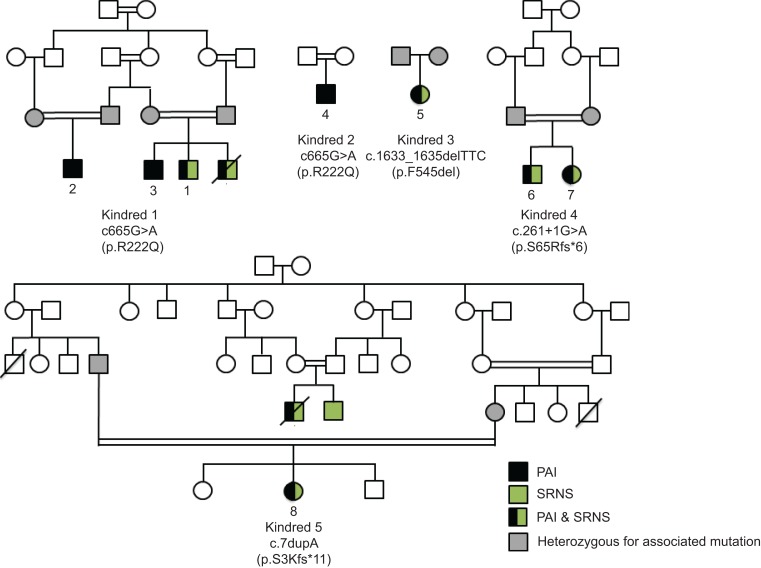
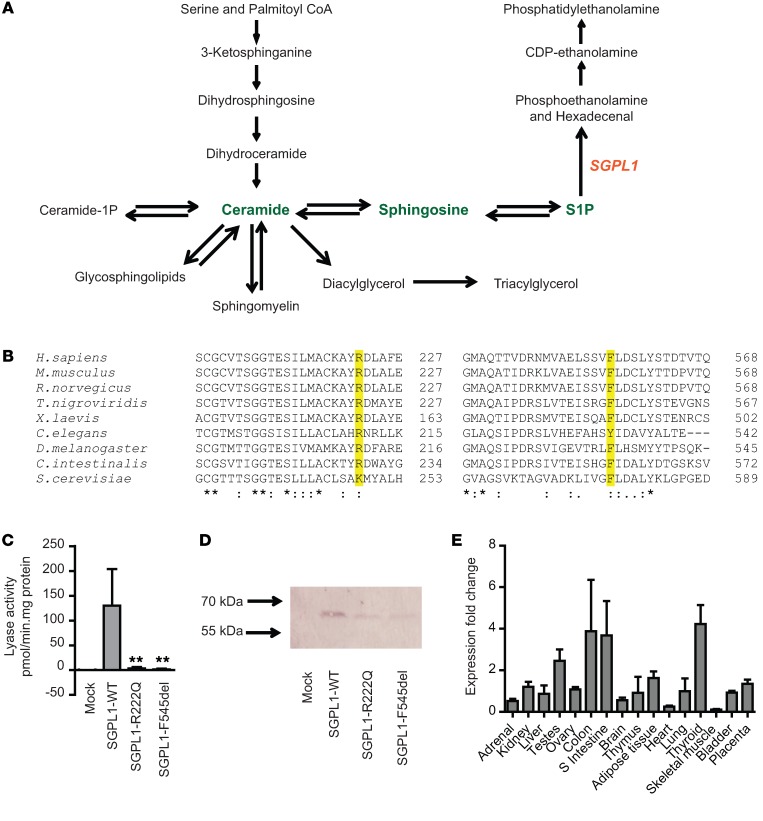
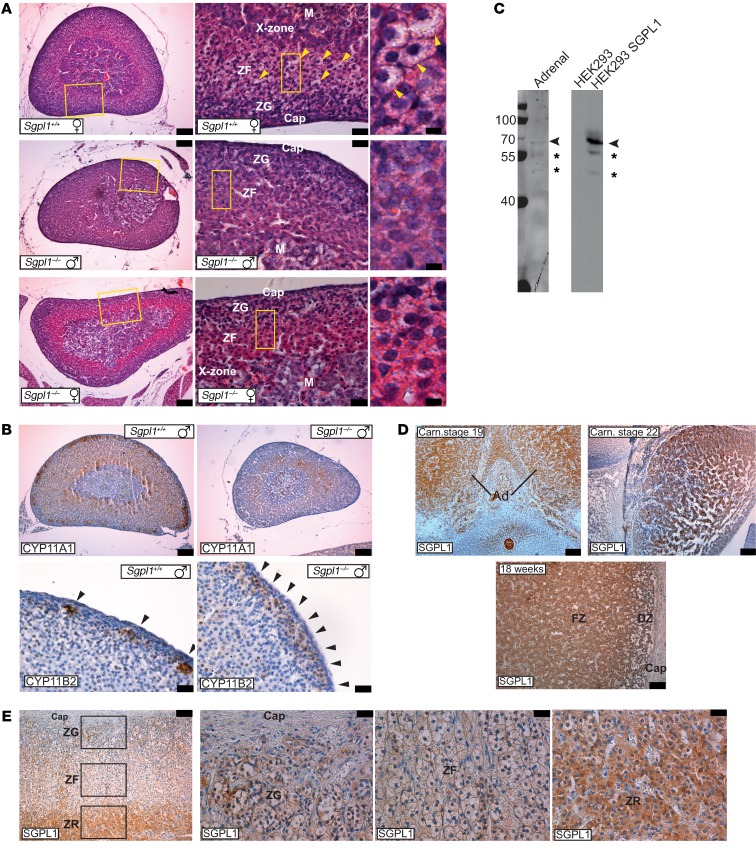
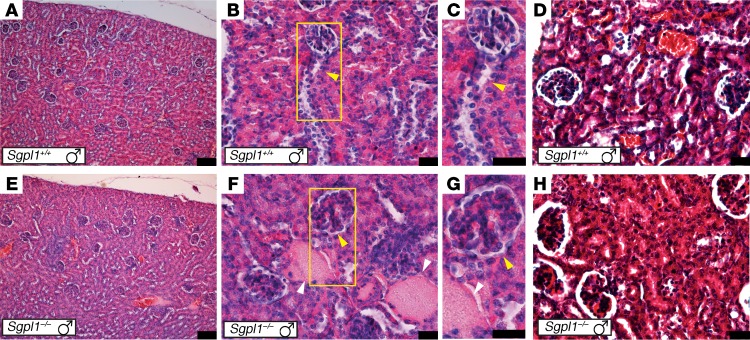
Primary adrenal insufficiency is life threatening and can present alone or in combination with other comorbidities. Here, we have described a primary adrenal insufficiency syndrome and steroid-resistant nephrotic syndrome caused by loss-of-function mutations in sphingosine-1-phosphate lyase (SGPL1). SGPL1 executes the final decisive step of the sphingolipid breakdown pathway, mediating the irreversible cleavage of the lipid-signaling molecule sphingosine-1-phosphate (S1P). Mutations in other upstream components of the pathway lead to harmful accumulation of lysosomal sphingolipid species, which are associated with a series of conditions known as the sphingolipidoses. In this work, we have identified 4 different homozygous mutations, c.665G>A (p.R222Q), c.1633_1635delTTC (p.F545del), c.261+1G>A (p.S65Rfs*6), and c.7dupA (p.S3Kfs*11), in 5 families with the condition. In total, 8 patients were investigated, some of whom also manifested other features, including ichthyosis, primary hypothyroidism, neurological symptoms, and cryptorchidism. Sgpl1-/- mice recapitulated the main characteristics of the human disease with abnormal adrenal and renal morphology. Sgpl1-/- mice displayed disrupted adrenocortical zonation and defective expression of steroidogenic enzymes as well as renal histology in keeping with a glomerular phenotype. In summary, we have identified SGPL1 mutations in humans that perhaps represent a distinct multisystemic disorder of sphingolipid metabolism.
Conflict of interest statement
Figures
Comment in
-
Genetics: SGPL1 mutations cause a novel SRNS syndrome.Nat Rev Nephrol. 2017 Apr;13(4):191. doi: 10.1038/nrneph.2017.19. Epub 2017 Feb 20. Nat Rev Nephrol. 2017. PMID: 28218265 No abstract available.
Similar articles
-
Factors influencing survival in sphingosine phosphate lyase insufficiency syndrome: a retrospective cross-sectional natural history study of 76 patients.Orphanet J Rare Dis. 2024 Sep 27;19(1):355. doi: 10.1186/s13023-024-03311-w. Orphanet J Rare Dis. 2024. PMID: 39334450 Free PMC article.
-
Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications.Hum Mutat. 2017 Apr;38(4):365-372. doi: 10.1002/humu.23192. Epub 2017 Mar 6. Hum Mutat. 2017. PMID: 28181337 Free PMC article.
-
Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency.J Clin Invest. 2017 Mar 1;127(3):912-928. doi: 10.1172/JCI89626. Epub 2017 Feb 6. J Clin Invest. 2017. PMID: 28165339 Free PMC article.
-
A rare cause of nephrotic syndrome-sphingosine-1-phosphate lyase (SGPL1) deficiency: 6 cases and a review of the literature.Pediatr Nephrol. 2023 Mar;38(3):711-719. doi: 10.1007/s00467-022-05656-5. Epub 2022 Jun 24. Pediatr Nephrol. 2023. PMID: 35748945 Review.
-
Congenital adrenal calcifications as the first clinical indication of sphingosine lyase insufficiency syndrome: A case report and review of the literature.Am J Med Genet A. 2022 Nov;188(11):3312-3317. doi: 10.1002/ajmg.a.62956. Epub 2022 Aug 16. Am J Med Genet A. 2022. PMID: 35972040 Free PMC article. Review.
Cited by
-
Kidney derived apolipoprotein M and its role in acute kidney injury.Front Pharmacol. 2024 Jan 19;15:1328259. doi: 10.3389/fphar.2024.1328259. eCollection 2024. Front Pharmacol. 2024. PMID: 38313311 Free PMC article.
-
Factors influencing survival in sphingosine phosphate lyase insufficiency syndrome: a retrospective cross-sectional natural history study of 76 patients.Orphanet J Rare Dis. 2024 Sep 27;19(1):355. doi: 10.1186/s13023-024-03311-w. Orphanet J Rare Dis. 2024. PMID: 39334450 Free PMC article.
-
Adrenal Dysfunction in Mitochondrial Diseases.Int J Mol Sci. 2023 Jan 6;24(2):1126. doi: 10.3390/ijms24021126. Int J Mol Sci. 2023. PMID: 36674647 Free PMC article. Review.
-
Combined novel homozygous variants in both SGPL1 and STAT1 presenting with severe combined immune deficiency: case report and literature review.Front Immunol. 2023 Jun 12;14:1186575. doi: 10.3389/fimmu.2023.1186575. eCollection 2023. Front Immunol. 2023. PMID: 37377976 Free PMC article. Review.
-
Identification of Glomerular and Plasma Apolipoprotein M as Novel Biomarkers in Glomerular Disease.Kidney Int Rep. 2023 Feb 4;8(4):884-897. doi: 10.1016/j.ekir.2023.01.031. eCollection 2023 Apr. Kidney Int Rep. 2023. PMID: 37069998 Free PMC article.
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
