

The GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy
- PMID: 27491214
- PMCID: PMC8186028
The GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy
Abstract
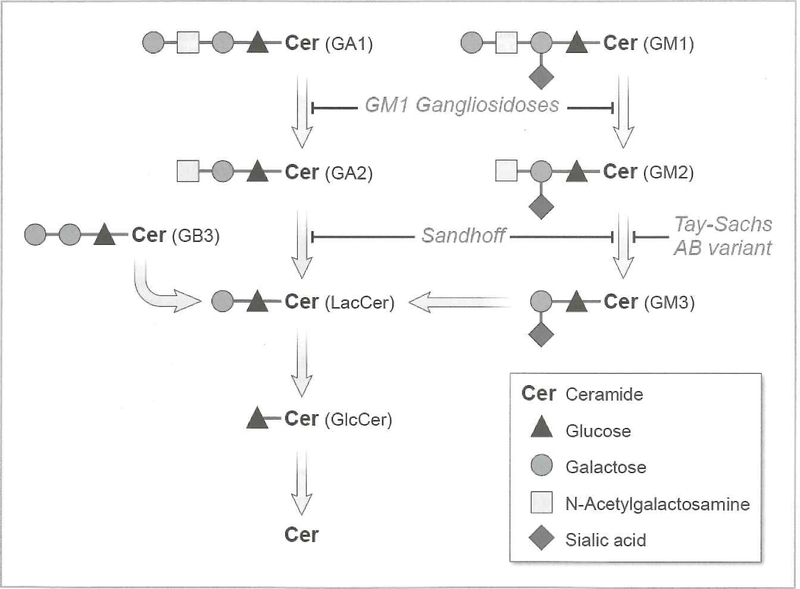
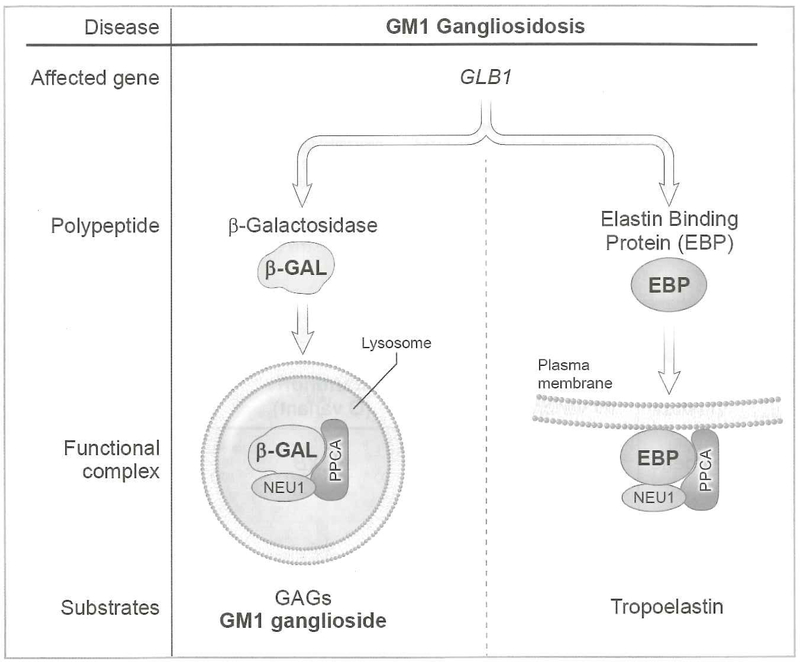
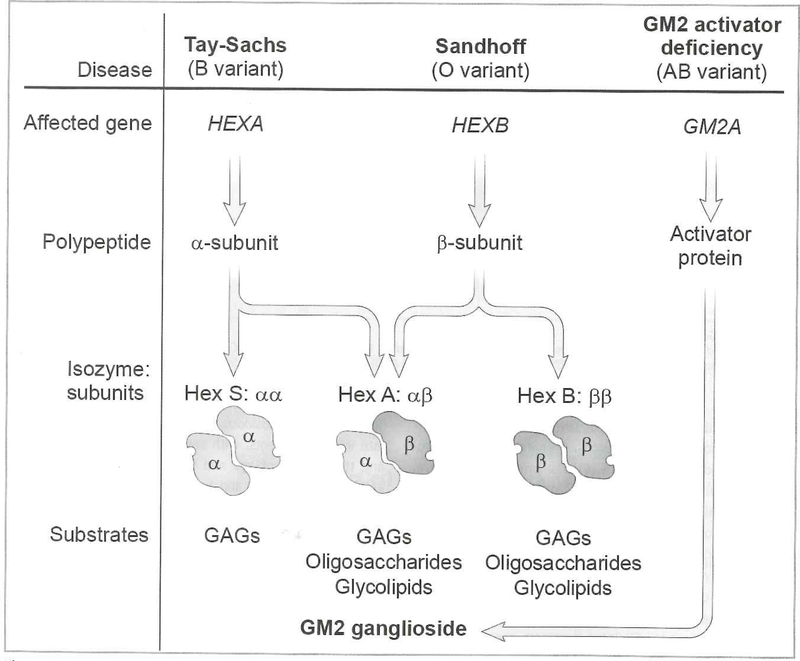
The gangliosidoses are lysosomal storage disorders caused by accumulation of GM1 or GM2 gangliosides. GM1 gangliosidosis has both central nervous system and systemic findings; while, GM2 gangliosidosis is restricted primarily to the central nervous system. Both disorders have autosomal recessive modes of inheritance and a continuum of clinical presentations from a severe infantile form to a milder, chronic adult form. Both are devastating diseases without cure or specific treatment however, with the use of supportive aggressive medical management, the lifespan and quality of life has been extended for both diseases. Naturally occurring and engineered animal models that mimic the human diseases have enhanced our understanding of the pathogenesis of disease progression. Some models have shown significant improvement in symptoms and lifespan with enzyme replacement, substrate reduction, and anti-inflammatory treatments alone or in combination. More recently gene therapy has shown impressive results in large and small animal models. Treatment with FDA-approved glucose analogs to reduce the amount of ganglioside substrate is used as off-label treatments for some patients. Therapies also under clinical development include small molecule chaperones and gene therapy.
Conflict of interest statement
Disclosure
The authors have no conflicts of interest to declare
Figures
Similar articles
-
Infantile gangliosidoses: Mapping a timeline of clinical changes.Mol Genet Metab. 2017 Jun;121(2):170-179. doi: 10.1016/j.ymgme.2017.04.011. Epub 2017 Apr 29. Mol Genet Metab. 2017. PMID: 28476546 Free PMC article. Clinical Trial.
-
Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis.J Neurosci. 2013 Jun 19;33(25):10195-208. doi: 10.1523/JNEUROSCI.0822-13.2013. J Neurosci. 2013. PMID: 23785136 Free PMC article. Review.
-
Distinct progression patterns of brain disease in infantile and juvenile gangliosidoses: Volumetric quantitative MRI study.Mol Genet Metab. 2018 Feb;123(2):97-104. doi: 10.1016/j.ymgme.2017.12.432. Epub 2017 Dec 20. Mol Genet Metab. 2018. PMID: 29352662 Free PMC article.
-
The GM2 gangliosidoses: Unlocking the mysteries of pathogenesis and treatment.Neurosci Lett. 2021 Nov 1;764:136195. doi: 10.1016/j.neulet.2021.136195. Epub 2021 Aug 25. Neurosci Lett. 2021. PMID: 34450229 Free PMC article. No abstract available.
-
[Molecular pathogenesis and therapeutic approach of GM2 gangliosidosis].Yakugaku Zasshi. 2013;133(2):269-74. doi: 10.1248/yakushi.12-00199. Yakugaku Zasshi. 2013. PMID: 23370522 Review. Japanese.
Cited by
-
Enhanced Stability of Long-Living Immobilized Recombinant β-d-N-Acetyl-Hexosaminidase A on Polylactic Acid (PLA) Films for Potential Biomedical Applications.J Funct Biomater. 2021 May 11;12(2):32. doi: 10.3390/jfb12020032. J Funct Biomater. 2021. PMID: 34064736 Free PMC article.
-
Rapid Identification of New Biomarkers for the Classification of GM1 Type 2 Gangliosidosis Using an Unbiased 1H NMR-Linked Metabolomics Strategy.Cells. 2021 Mar 5;10(3):572. doi: 10.3390/cells10030572. Cells. 2021. PMID: 33807817 Free PMC article.
-
Emptying the stores: lysosomal diseases and therapeutic strategies.Nat Rev Drug Discov. 2018 Feb;17(2):133-150. doi: 10.1038/nrd.2017.214. Epub 2017 Nov 17. Nat Rev Drug Discov. 2018. PMID: 29147032 Review.
-
Metabolic and genetic disorders mimicking cerebral palsy.Neurosciences (Riyadh). 2019 Jul;24(3):155-163. doi: 10.17712/nsj.2019.3.20190045. Neurosciences (Riyadh). 2019. PMID: 31380813 Free PMC article. Review.
-
Therapeutic advantages of combined gene/cell therapy strategies in a murine model of GM2 gangliosidosis.Mol Ther Methods Clin Dev. 2022 Mar 16;25:170-189. doi: 10.1016/j.omtm.2022.03.011. eCollection 2022 Jun 9. Mol Ther Methods Clin Dev. 2022. PMID: 35434178 Free PMC article.
References
-
- Regier DS, Tifft CJ. GLB1-Related Disorders. In: GeneReviews(R). Edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Smith RJH, Stephens K. Seattle (WA) 2013
-
- Regier DS, Oetgen M, Tanpaiboon P. Mucopolysaccharidosis Type IVA. In: GeneReviews(R). Edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K. Seattle (WA) 2015
-
- Schulze H, Sandhoff K. Sphingolipids and lysosomal pathologies. Biochim Biophys Acta 2014;1841:799–810 - PubMed
-
- Hofer D, Paul K, Fantur K, Beck M, Roubergue A, Vellodi A, Poorthuis BJ, Michelakakis H, Plecko B, Paschke E. Phenotype determining alleles in GM1 gangliosidosis patients bearing novel GLB1 mutations. Clinical Genetics 2010;78:236–246 - PubMed
-
- Caciotti A, Garman SC, Rivera-Colon Y, Procopio E, Catarzi S, Ferri L, Guido C, Martelli P, Parini R, Antuzzi D, Battini R, Sibilio M, Simonati A, Fontana E, Salviati A, Akinci G, Cereda C, Dionisi-Vici C, Deodato F, d’Amico A, d’Azzo A, Bertini E, Filocamo M, Scarpa M, di Rocco M, Tifft CJ, Ciani F, Gasperini S, Pasquini E, Guerrini R, Donati AAA, Morrone A. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochimica et Biophysica Acta 2011;1812:782–790 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical