

Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss
- PMID: 26969326
- PMCID: PMC4796320
- DOI: 10.1007/s00439-016-1648-8
Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss
Abstract
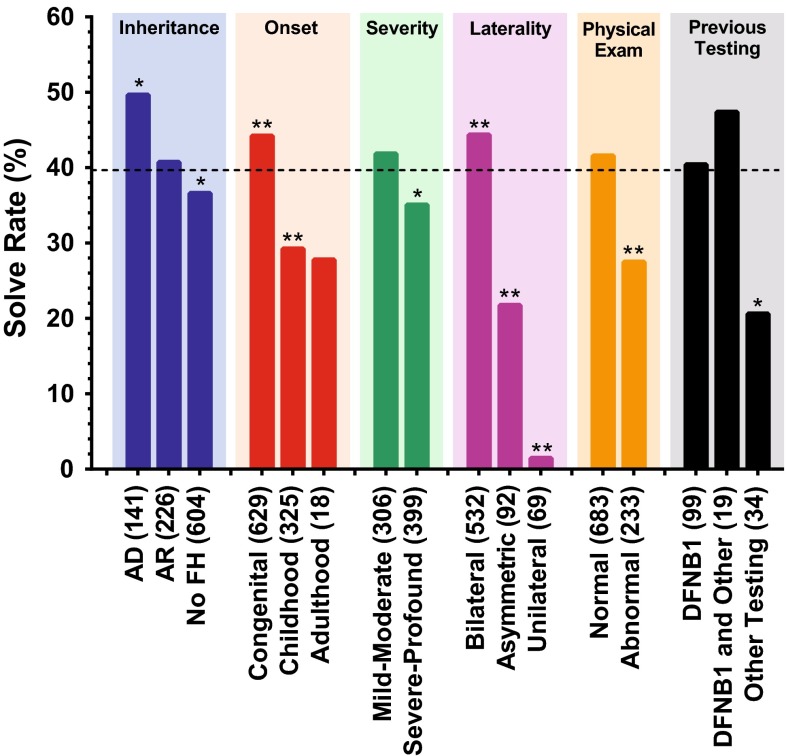
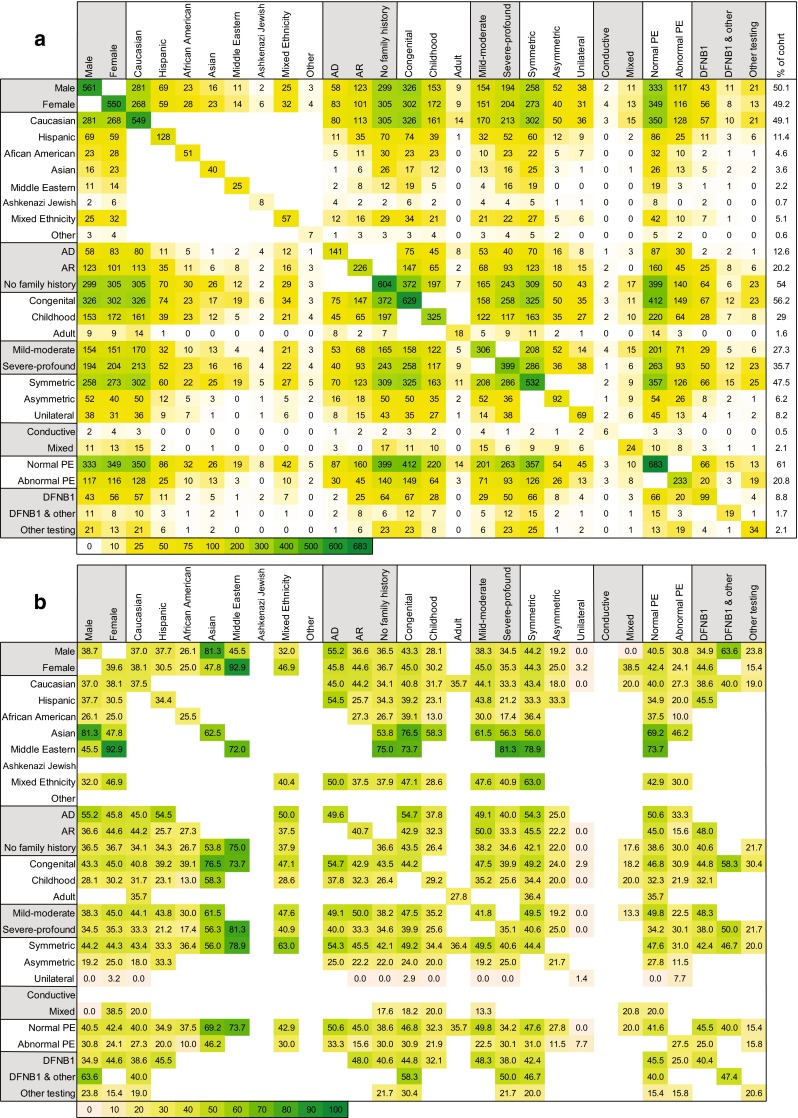
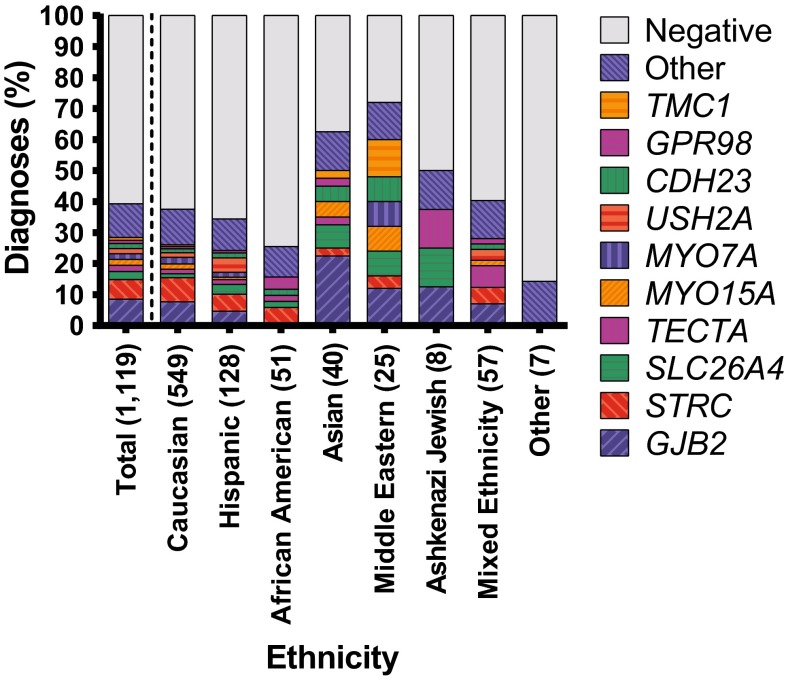
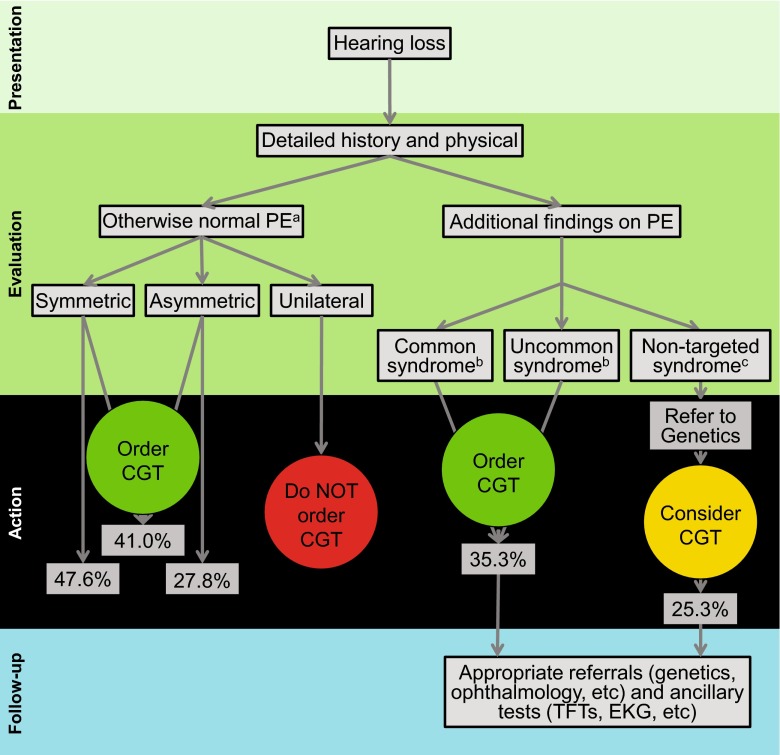
Hearing loss is the most common sensory deficit in humans, affecting 1 in 500 newborns. Due to its genetic heterogeneity, comprehensive diagnostic testing has not previously been completed in a large multiethnic cohort. To determine the aggregate contribution inheritance makes to non-syndromic hearing loss, we performed comprehensive clinical genetic testing with targeted genomic enrichment and massively parallel sequencing on 1119 sequentially accrued patients. No patient was excluded based on phenotype, inheritance or previous testing. Testing resulted in identification of the underlying genetic cause for hearing loss in 440 patients (39%). Pathogenic variants were found in 49 genes and included missense variants (49%), large copy number changes (18%), small insertions and deletions (18%), nonsense variants (8%), splice-site alterations (6%), and promoter variants (<1%). The diagnostic rate varied considerably based on phenotype and was highest for patients with a positive family history of hearing loss or when the loss was congenital and symmetric. The spectrum of implicated genes showed wide ethnic variability. These findings support the more efficient utilization of medical resources through the development of evidence-based algorithms for the diagnosis of hearing loss.
Figures
Similar articles
-
Genetic testing for sporadic hearing loss using targeted massively parallel sequencing identifies 10 novel mutations.Clin Genet. 2015 Jun;87(6):588-93. doi: 10.1111/cge.12431. Epub 2014 Aug 7. Clin Genet. 2015. PMID: 24853665
-
Navigating genetic diagnostics in patients with hearing loss.Curr Opin Pediatr. 2016 Dec;28(6):705-712. doi: 10.1097/MOP.0000000000000410. Curr Opin Pediatr. 2016. PMID: 27552069 Free PMC article. Review.
-
Genomic and phenotypic landscapes of X-linked hereditary hearing loss in the Chinese population.Orphanet J Rare Dis. 2024 Sep 13;19(1):342. doi: 10.1186/s13023-024-03338-z. Orphanet J Rare Dis. 2024. PMID: 39272213 Free PMC article.
-
Investigation of Targeted Genes and Identification of Novel Variants with Next Generation Sequencing Method in Hearing Loss.J Int Adv Otol. 2024 Jul 29;20(4):312-324. doi: 10.5152/iao.2024.22919. J Int Adv Otol. 2024. PMID: 39161163 Free PMC article.
-
Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss: The New Standard of Care.Otolaryngol Head Neck Surg. 2015 Aug;153(2):175-82. doi: 10.1177/0194599815591156. Epub 2015 Jun 17. Otolaryngol Head Neck Surg. 2015. PMID: 26084827 Free PMC article. Review.
Cited by
-
TSPEAR variants are primarily associated with ectodermal dysplasia and tooth agenesis but not hearing loss: A novel cohort study.Am J Med Genet A. 2021 Aug;185(8):2417-2433. doi: 10.1002/ajmg.a.62347. Epub 2021 May 27. Am J Med Genet A. 2021. PMID: 34042254 Free PMC article.
-
A novel KCNQ4 gene variant (c.857A>G; p.Tyr286Cys) in an extended family with non‑syndromic deafness 2A.Mol Med Rep. 2021 Jun;23(6):420. doi: 10.3892/mmr.2021.12059. Epub 2021 Apr 13. Mol Med Rep. 2021. PMID: 33846771 Free PMC article.
-
Heterogeneity of Hereditary Hearing Loss in Iran: a Comprehensive Review.Arch Iran Med. 2016 Oct 1;19(10):720-728. Arch Iran Med. 2016. PMID: 27743438 Free PMC article. Review.
-
A RIPOR2 in-frame deletion is a frequent and highly penetrant cause of adult-onset hearing loss.J Med Genet. 2020 Jul 6:jmedgenet-2020-106863. doi: 10.1136/jmedgenet-2020-106863. Online ahead of print. J Med Genet. 2020. PMID: 32631815 Free PMC article.
-
Four Novel Variants in POU4F3 Cause Autosomal Dominant Nonsyndromic Hearing Loss.Neural Plast. 2020 Jul 1;2020:6137083. doi: 10.1155/2020/6137083. eCollection 2020. Neural Plast. 2020. PMID: 32684921 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
