

The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome
- PMID: 25758994
- PMCID: PMC4795191
- DOI: 10.1038/ejhg.2015.32
The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome
Abstract
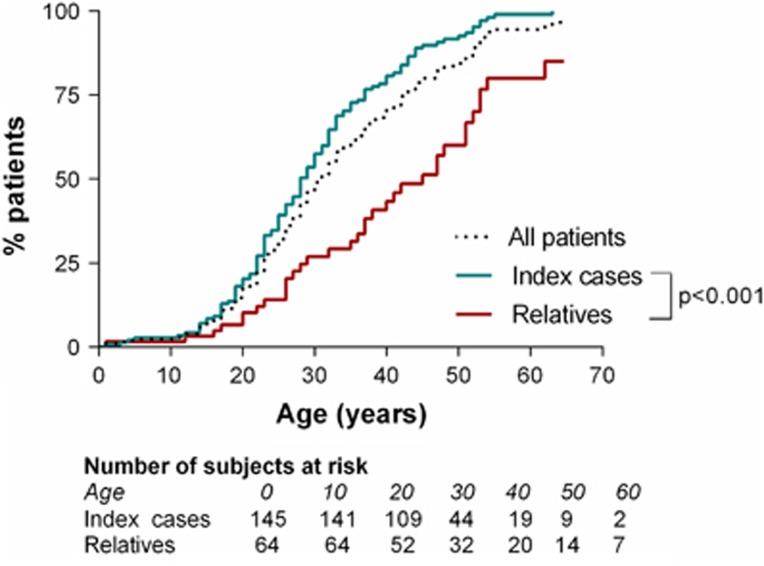
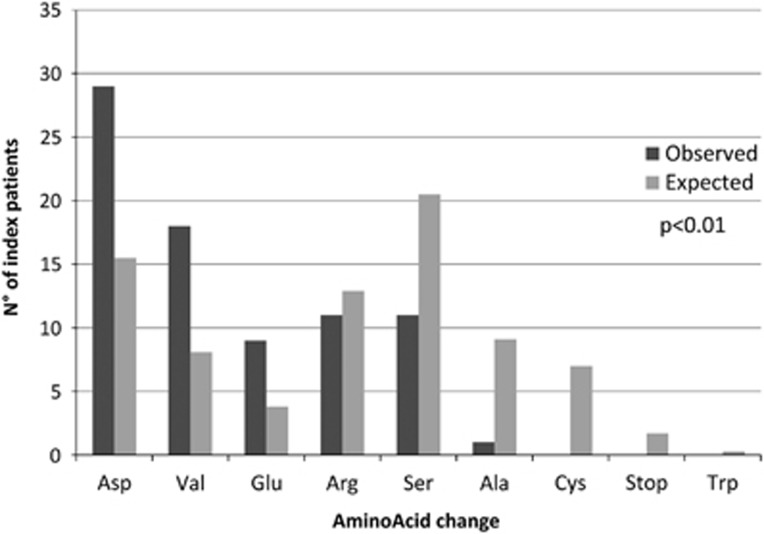
Vascular Ehlers-Danlos syndrome (vEDS) is a rare and severe autosomal dominant disorder caused by variants at the COL3A1 gene. Clinical characteristics and course of disease of 215 molecularly proven patients (146 index cases and 69 relatives) were analysed. We found 126 distincts variants that were divided into five groups: (1) Glycine substitutions (n=71), (2) splice-site and in-frame insertions-deletions (n=36), (3) variants leading to haplo-insufficiency (n=7), (4) non-glycine missense variants within the triple helix (n=4 variants), and (5) non-glycine missense variants or in-frame insertions-deletions, in the N- or C-terminal part of the protein (n=8). Overall, our cohort confirmed the severity of the disease with a median age at first complication of 29 years (IQR 22-39), the most frequent being arterial (48%) and digestive (24%) ruptures. Groups 2 and 1 were significantly more severe than groups 3-5, with extreme median ages at first major complication of 23-47 years. Patients of groups 3-5 had a less typical phenotype and remarkably absence of digestive events. The distribution of glycine-replacing amino acids was strongly biased towards more destabilizing residues of the collagen assembly. Thus the natural course of vEDS and the clinical phenotype of patients are influenced by the type of COL3A1 variant. This study also confirms that patients with variants located in the C- and N-termini or leading to haplo-insufficiency have milder course of the disease and less prevalent diagnostic criteria. These findings may help refine diagnostic strategy, genetic counselling and clinical care.
Figures
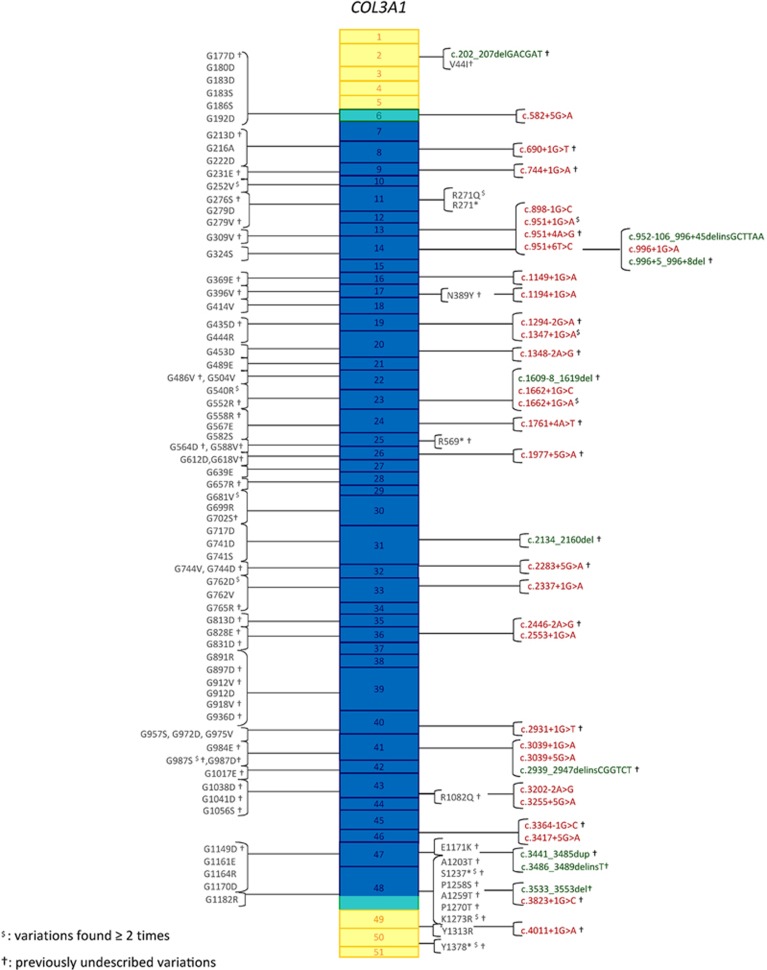
 indicates the variants found more than once in the index cases cohort; †indicates previously undescribed variants.
indicates the variants found more than once in the index cases cohort; †indicates previously undescribed variants.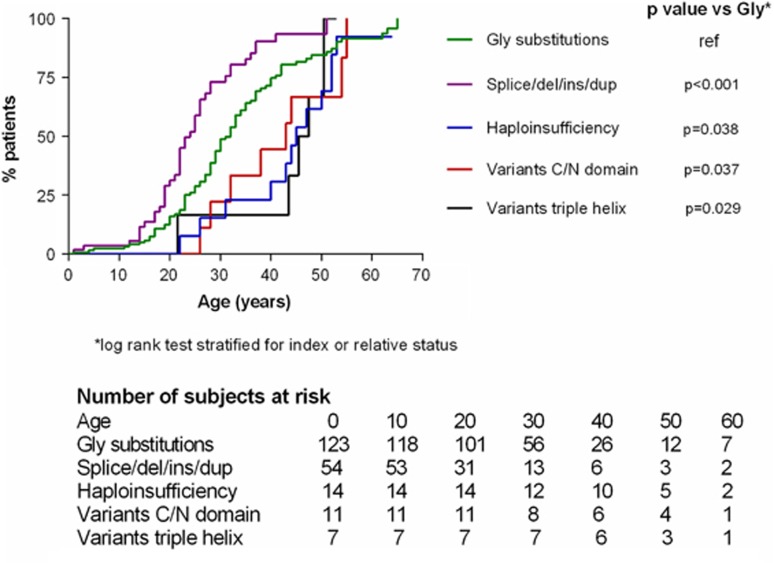
Similar articles
-
A multi-institutional experience in the aortic and arterial pathology in individuals with genetically confirmed vascular Ehlers-Danlos syndrome.J Vasc Surg. 2019 Nov;70(5):1543-1554. doi: 10.1016/j.jvs.2019.01.069. Epub 2019 May 21. J Vasc Surg. 2019. PMID: 31126764 Free PMC article.
-
Transcriptome analysis of skin fibroblasts with dominant negative COL3A1 mutations provides molecular insights into the etiopathology of vascular Ehlers-Danlos syndrome.PLoS One. 2018 Jan 18;13(1):e0191220. doi: 10.1371/journal.pone.0191220. eCollection 2018. PLoS One. 2018. PMID: 29346445 Free PMC article.
-
Atypical COL3A1 variants (glutamic acid to lysine) cause vascular Ehlers-Danlos syndrome with a consistent phenotype of tissue fragility and skin hyperextensibility.Genet Med. 2019 Sep;21(9):2081-2091. doi: 10.1038/s41436-019-0470-9. Epub 2019 Mar 6. Genet Med. 2019. PMID: 30837697
-
Vascular Ehlers-Danlos Syndrome With a Novel Missense COL3A1 Mutation Present With Pulmonary Complications and Iliac Arterial Dissection.Vasc Endovascular Surg. 2018 Feb;52(2):138-142. doi: 10.1177/1538574417745432. Epub 2017 Dec 7. Vasc Endovascular Surg. 2018. PMID: 29216800 Review.
-
A Novel Frameshift COL3A1 Variant in Vascular Ehlers-Danlos Syndrome.Ann Vasc Surg. 2019 Nov;61:472.e9-472.e13. doi: 10.1016/j.avsg.2019.05.057. Epub 2019 Aug 5. Ann Vasc Surg. 2019. PMID: 31394236 Review.
Cited by
-
Assessment of arterial damage in vascular Ehlers-Danlos syndrome: A retrospective multicentric cohort.Front Cardiovasc Med. 2022 Oct 3;9:953894. doi: 10.3389/fcvm.2022.953894. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 36262204 Free PMC article.
-
The Ehlers-Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism.Genes (Basel). 2022 Jan 29;13(2):265. doi: 10.3390/genes13020265. Genes (Basel). 2022. PMID: 35205310 Free PMC article. Review.
-
Case 1/2019 - A 51-year-old Man with Arterial Hypertension, Aortic Dissection and Aortic Valve Regurgitation, in Addition to Heart Failure with Unchanged Clinical Course After Surgical Intervention.Arq Bras Cardiol. 2019 Feb;112(2):204-210. doi: 10.5935/abc.20190013. Arq Bras Cardiol. 2019. PMID: 30785587 Free PMC article. No abstract available.
-
Extracutaneous features and complications of the Ehlers-Danlos syndromes: A systematic review.Front Med (Lausanne). 2023 Jan 23;10:1053466. doi: 10.3389/fmed.2023.1053466. eCollection 2023. Front Med (Lausanne). 2023. PMID: 36756177 Free PMC article.
-
Recurrent spontaneous small bowel perforations with a rare pathology: non-familial visceral myopathy.BMJ Case Rep. 2021 May 13;14(5):e240923. doi: 10.1136/bcr-2020-240923. BMJ Case Rep. 2021. PMID: 33986007 Free PMC article.
References
-
- Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G, Richards AJ: COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol 1996; 135: 163–181. - PubMed
-
- Pepin M, Schwarze U, Superti-Furga A, Byers PH: Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673–680. - PubMed
-
- Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ: Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 1998; 77: 31–37. - PubMed
-
- Leistritz DF, Pepin MG, Schwarze U, Byers PH: COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med 2011; 13: 717–722. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
