

Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios
- PMID: 25590979
- PMCID: PMC4791490
- DOI: 10.1038/gim.2014.191
Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios
Abstract
Purpose: Despite the recognized clinical value of exome-based diagnostics, methods for comprehensive genomic interpretation remain immature. Diagnoses are based on known or presumed pathogenic variants in genes already associated with a similar phenotype. Here, we extend this paradigm by evaluating novel bioinformatics approaches to aid identification of new gene-disease associations.
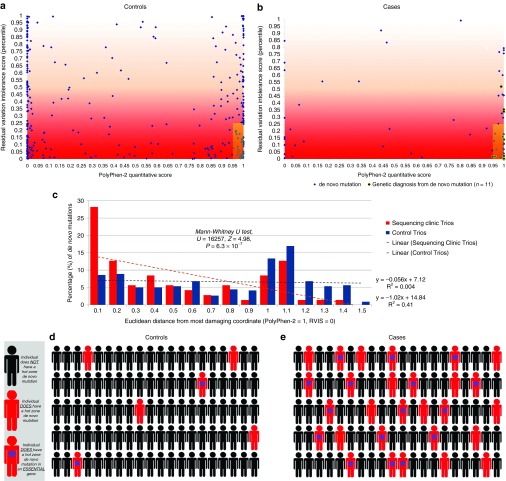
Methods: We analyzed 119 trios to identify both diagnostic genotypes in known genes and candidate genotypes in novel genes. We considered qualifying genotypes based on their population frequency and in silico predicted effects we also characterized the patterns of genotypes enriched among this collection of patients.
Results: We obtained a genetic diagnosis for 29 (24%) of our patients. We showed that patients carried an excess of damaging de novo mutations in intolerant genes, particularly those shown to be essential in mice (P = 3.4 × 10(-8)). This enrichment is only partially explained by mutations found in known disease-causing genes.
Conclusion: This work indicates that the application of appropriate bioinformatics analyses to clinical sequence data can also help implicate novel disease genes and suggest expanded phenotypes for known disease genes. These analyses further suggest that some cases resolved by whole-exome sequencing will have direct therapeutic implications.
Figures
Similar articles
-
Lessons learned from additional research analyses of unsolved clinical exome cases.Genome Med. 2017 Mar 21;9(1):26. doi: 10.1186/s13073-017-0412-6. Genome Med. 2017. PMID: 28327206 Free PMC article.
-
Phenotype-driven variant filtration strategy in exome sequencing toward a high diagnostic yield and identification of 85 novel variants in 400 patients with rare Mendelian disorders.Am J Med Genet A. 2021 Aug;185(8):2561-2571. doi: 10.1002/ajmg.a.62338. Epub 2021 May 19. Am J Med Genet A. 2021. PMID: 34008892
-
Computational and statistical approaches to analyzing variants identified by exome sequencing.Genome Biol. 2011 Sep 14;12(9):227. doi: 10.1186/gb-2011-12-9-227. Genome Biol. 2011. PMID: 21920052 Free PMC article. Review.
-
Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases.Genet Med. 2017 Feb;19(2):224-235. doi: 10.1038/gim.2016.95. Epub 2016 Aug 11. Genet Med. 2017. PMID: 27513193 Free PMC article.
-
Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care.Clin Genet. 2016 Mar;89(3):275-84. doi: 10.1111/cge.12654. Epub 2015 Sep 22. Clin Genet. 2016. PMID: 26283276 Free PMC article. Review.
Cited by
-
Phenotype-driven approaches to enhance variant prioritization and diagnosis of rare disease.Hum Mutat. 2022 Aug;43(8):1071-1081. doi: 10.1002/humu.24380. Epub 2022 Apr 27. Hum Mutat. 2022. PMID: 35391505 Free PMC article. Review.
-
Encoding Clinical Data with the Human Phenotype Ontology for Computational Differential Diagnostics.Curr Protoc Hum Genet. 2019 Sep;103(1):e92. doi: 10.1002/cphg.92. Curr Protoc Hum Genet. 2019. PMID: 31479590 Free PMC article.
-
Novel detection of mutation in the TECPR2 gene in a Chinese hereditary spastic paraplegia 49 patient: a case report.BMC Neurol. 2022 Feb 7;22(1):47. doi: 10.1186/s12883-022-02572-x. BMC Neurol. 2022. PMID: 35130874 Free PMC article.
-
Case Report: A Relatively Mild Phenotype Produced by Novel Mutations in the SEPSECS Gene.Front Pediatr. 2022 Jan 26;9:805575. doi: 10.3389/fped.2021.805575. eCollection 2021. Front Pediatr. 2022. PMID: 35155316 Free PMC article.
-
Interest of exome sequencing trio-like strategy based on pooled parental DNA for diagnosis and translational research in rare diseases.Mol Genet Genomic Med. 2021 Dec;9(12):e1836. doi: 10.1002/mgg3.1836. Epub 2021 Oct 30. Mol Genet Genomic Med. 2021. PMID: 34716697 Free PMC article.
References
-
- de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–1929. - PubMed
Publication types
MeSH terms
Grants and funding
- UO1AIO67854/PHS HHS/United States
- RCMH089905/PHS HHS/United States
- U01-NS077303/NS/NINDS NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- P30 AG028377/AG/NIA NIH HHS/United States
- R01MH077139/MH/NIMH NIH HHS/United States
- T32 GM007754/GM/NIGMS NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- R01 MH077139/MH/NIMH NIH HHS/United States
- K01 MH098126/MH/NIMH NIH HHS/United States
- R01MH099216/MH/NIMH NIH HHS/United States
- RC2 MH089915/MH/NIMH NIH HHS/United States
- RC2MH089915/MH/NIMH NIH HHS/United States
- HL-102924/HL/NHLBI NIH HHS/United States
- U01 NS053998/NS/NINDS NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- R01MH097971/MH/NIMH NIH HHS/United States
- R01 MH097971/MH/NIMH NIH HHS/United States
- R25 GM086262/GM/NIGMS NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- U01 NS077303/NS/NINDS NIH HHS/United States
- U19 AI067854/AI/NIAID NIH HHS/United States
- R01 MH099216/MH/NIMH NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- K01MH098126/MH/NIMH NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- AG-NS-0441-08/AG/NIA NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
- U01-NS053998/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
