

Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E
- PMID: 25033457
- PMCID: PMC4164503
- DOI: 10.4161/epi.29676
Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E
Abstract
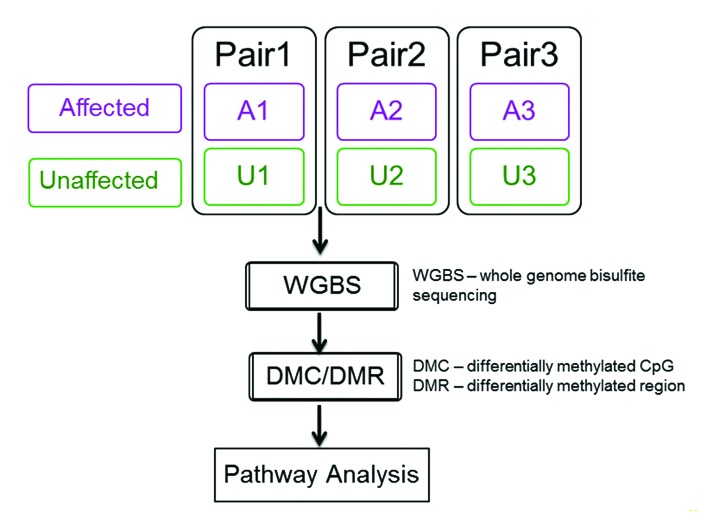
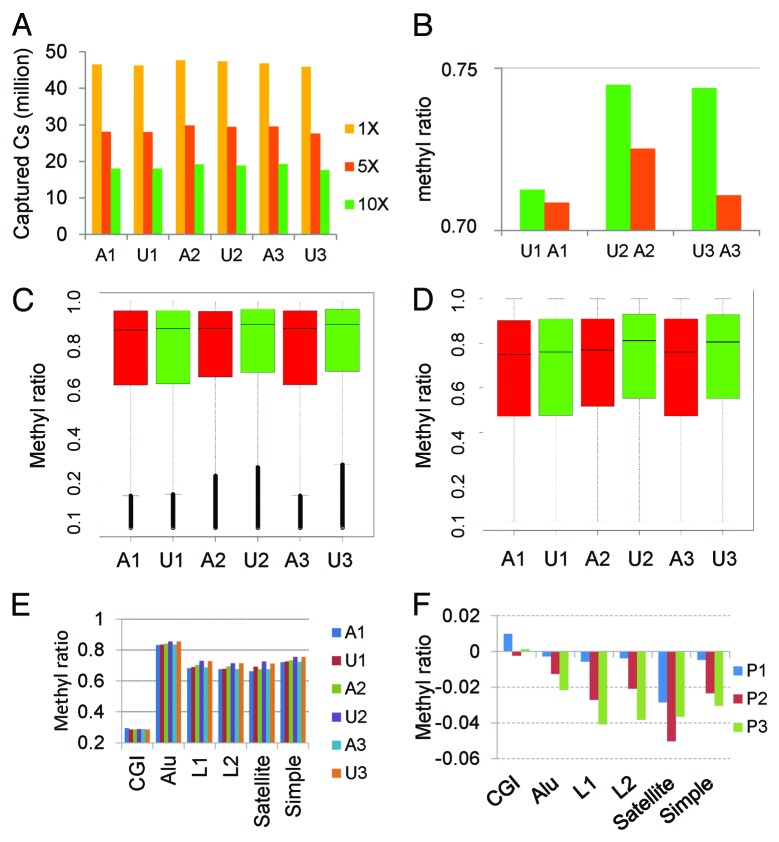
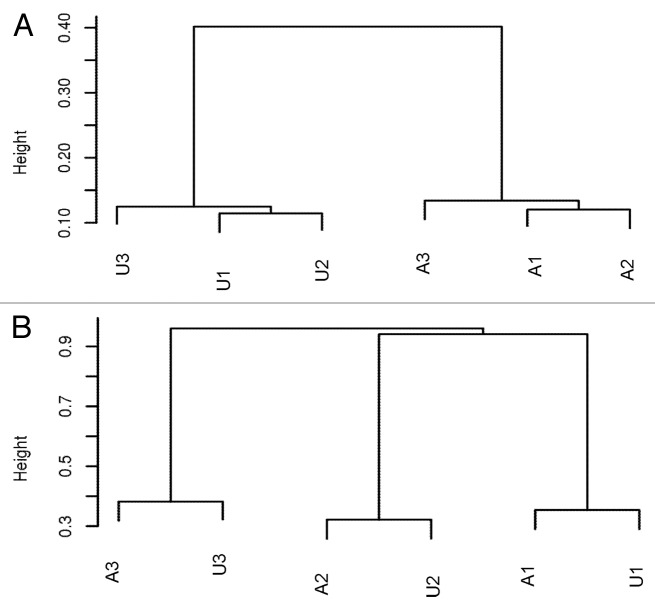
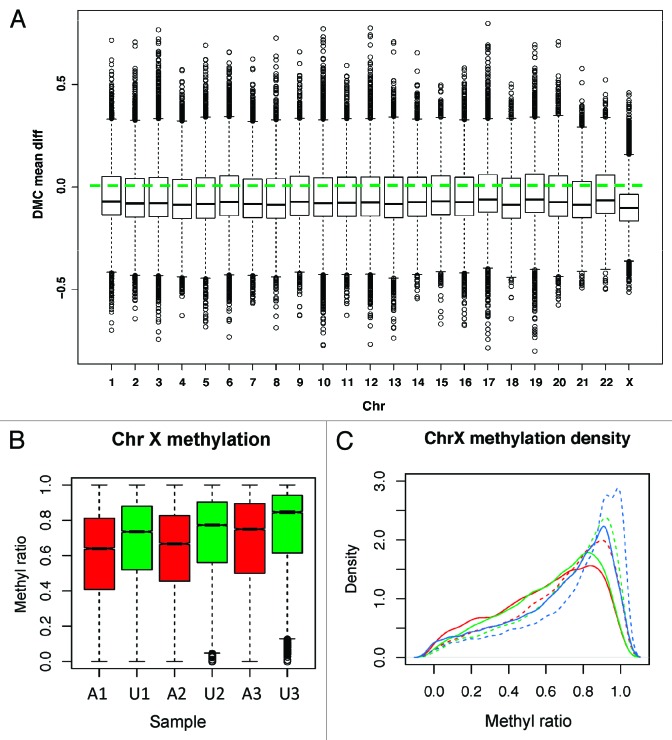
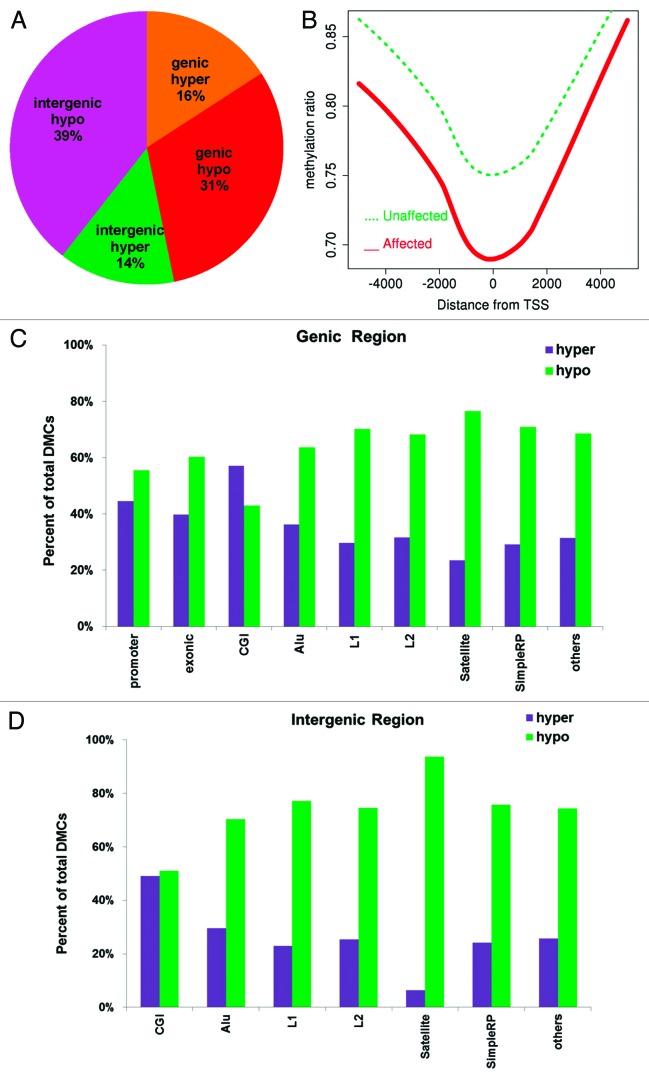
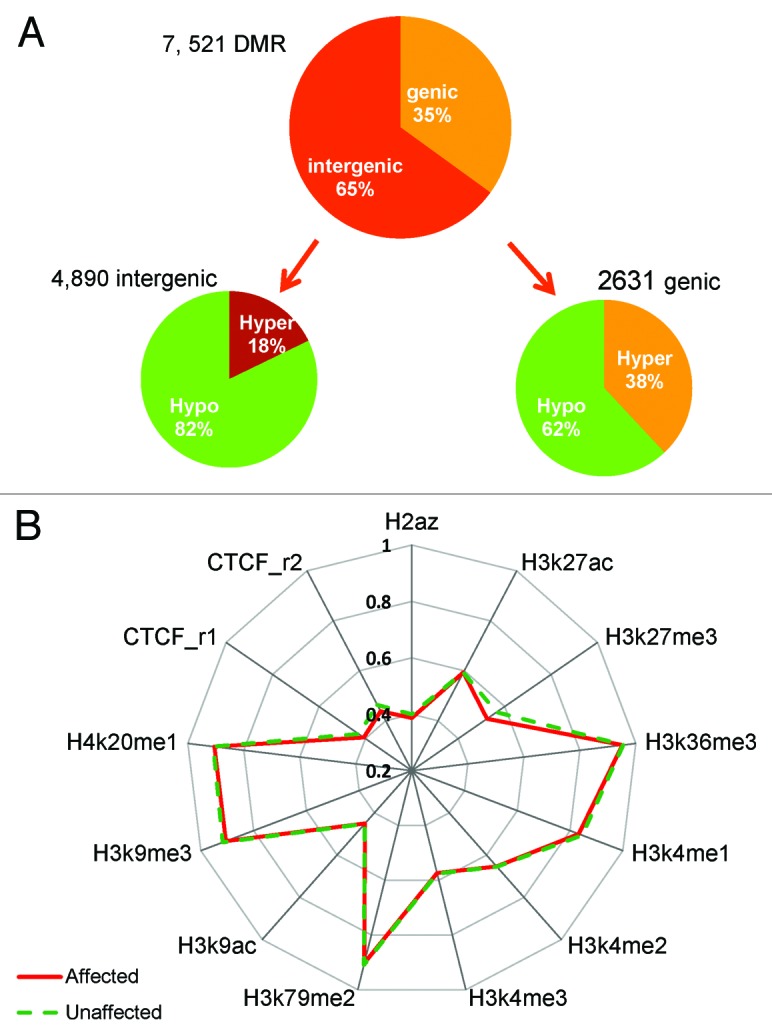
DNA methyltransferase 1 (DNMT1) is essential for DNA methylation, gene regulation and chromatin stability. We previously discovered DNMT1 mutations cause hereditary sensory and autonomic neuropathy type 1 with dementia and hearing loss (HSAN1E; OMIM 614116). HSAN1E is the first adult-onset neurodegenerative disorder caused by a defect in a methyltransferase gene. HSAN1E patients appear clinically normal until young adulthood, then begin developing the characteristic symptoms involving central and peripheral nervous systems. Some HSAN1E patients also develop narcolepsy and it has recently been suggested that HSAN1E is allelic to autosomal dominant cerebellar ataxia, deafness, with narcolepsy (ADCA-DN; OMIM 604121), which is also caused by mutations in DNMT1. A hotspot mutation Y495C within the targeting sequence domain of DNMT1 has been identified among HSAN1E patients. The mutant DNMT1 protein shows premature degradation and reduced DNA methyltransferase activity. Herein, we investigate genome-wide DNA methylation at single-base resolution through whole-genome bisulfite sequencing of germline DNA in 3 pairs of HSAN1E patients and their gender- and age-matched siblings. Over 1 billion 75-bp single-end reads were generated for each sample. In the 3 affected siblings, overall methylation loss was consistently found in all chromosomes with X and 18 being most affected. Paired sample analysis identified 564,218 differentially methylated CpG sites (DMCs; P<0.05), of which 300 134 were intergenic and 264 084 genic CpGs. Hypomethylation was predominant in both genic and intergenic regions, including promoters, exons, most CpG islands, L1, L2, Alu, and satellite repeats and simple repeat sequences. In some CpG islands, hypermethylated CpGs outnumbered hypomethylated CpGs. In 201 imprinted genes, there were more DMCs than in non-imprinted genes and most were hypomethylated. Differentially methylated region (DMR) analysis identified 5649 hypomethylated and 1872 hypermethylated regions. Importantly, pathway analysis revealed 1693 genes associated with the identified DMRs were highly associated in diverse neurological disorders and NAD+/NADH metabolism pathways is implicated in the pathogenesis. Our results provide novel insights into the epigenetic mechanism of neurodegeneration arising from a hotspot DNMT1 mutation and reveal pathways potentially important in a broad category of neurological and psychological disorders.
Keywords: DNA methylation; HSAN1E; OMIM 604121; OMIM 614116; neurodegeneration; whole genome bisulfite sequencing.
Figures
Similar articles
-
Defects of mutant DNMT1 are linked to a spectrum of neurological disorders.Brain. 2015 Apr;138(Pt 4):845-61. doi: 10.1093/brain/awv010. Epub 2015 Feb 11. Brain. 2015. PMID: 25678562 Free PMC article.
-
DNMT1 Y495C mutation interferes with maintenance methylation of imprinting control regions.Int J Biochem Cell Biol. 2024 Apr;169:106535. doi: 10.1016/j.biocel.2024.106535. Epub 2024 Jan 26. Int J Biochem Cell Biol. 2024. PMID: 38281697
-
A novel DNMT1 mutation associated with early onset hereditary sensory and autonomic neuropathy, cataplexy, cerebellar atrophy, scleroderma, endocrinopathy, and common variable immune deficiency.J Peripher Nerv Syst. 2016 Sep;21(3):150-3. doi: 10.1111/jns.12178. J Peripher Nerv Syst. 2016. PMID: 27277422
-
Effect of Disease-Associated Germline Mutations on Structure Function Relationship of DNA Methyltransferases.Genes (Basel). 2019 May 14;10(5):369. doi: 10.3390/genes10050369. Genes (Basel). 2019. PMID: 31091831 Free PMC article. Review.
-
Cerebellar Ataxia as a Common Clinical Presentation Associated with DNMT1 p.Y511H and a Review of the Literature.J Mol Neurosci. 2021 Sep;71(9):1796-1801. doi: 10.1007/s12031-020-01784-5. Epub 2021 Jan 12. J Mol Neurosci. 2021. PMID: 33433851 Review.
Cited by
-
Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis.Epigenomics. 2015 Aug;7(5):813-28. doi: 10.2217/epi.15.21. Epub 2015 Sep 14. Epigenomics. 2015. PMID: 26366945 Free PMC article. Review.
-
DNA methyltransferase 1 mutations and mitochondrial pathology: is mtDNA methylated?Front Genet. 2015 Mar 12;6:90. doi: 10.3389/fgene.2015.00090. eCollection 2015. Front Genet. 2015. PMID: 25815005 Free PMC article.
-
Vincristine-Induced Peripheral Neuropathy in Childhood Acute Lymphoblastic Leukemia: Genetic Variation as a Potential Risk Factor.Front Pharmacol. 2021 Dec 9;12:771487. doi: 10.3389/fphar.2021.771487. eCollection 2021. Front Pharmacol. 2021. PMID: 34955843 Free PMC article. Review.
-
DNMT1 mutations found in HSANIE patients affect interaction with UHRF1 and neuronal differentiation.Hum Mol Genet. 2017 Apr 15;26(8):1522-1534. doi: 10.1093/hmg/ddx057. Hum Mol Genet. 2017. PMID: 28334952 Free PMC article.
-
Stress and tinnitus.HNO. 2015 Apr;63(4):258-65. doi: 10.1007/s00106-014-2973-7. HNO. 2015. PMID: 25862619
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical