

Multiple phenotypes in phosphoglucomutase 1 deficiency
- PMID: 24499211
- PMCID: PMC4373661
- DOI: 10.1056/NEJMoa1206605
Multiple phenotypes in phosphoglucomutase 1 deficiency
Abstract
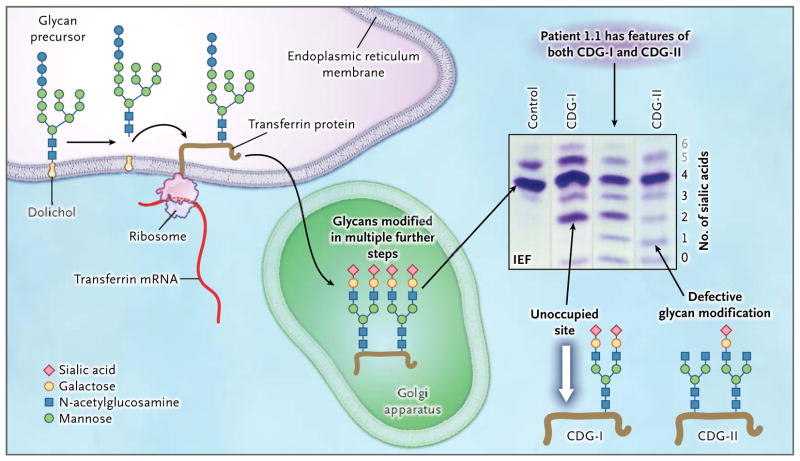
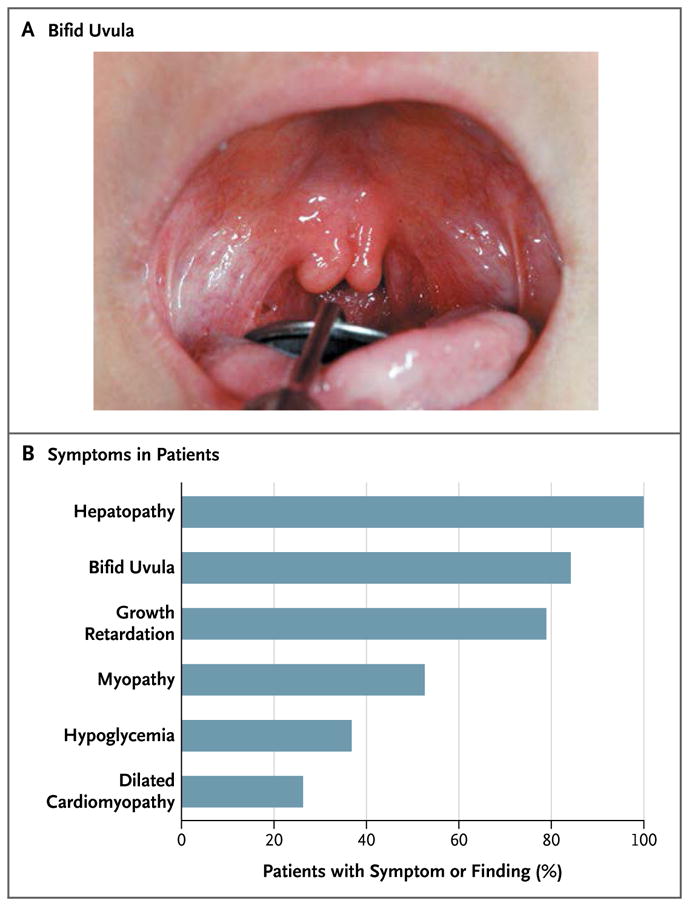
Background: Congenital disorders of glycosylation are genetic syndromes that result in impaired glycoprotein production. We evaluated patients who had a novel recessive disorder of glycosylation, with a range of clinical manifestations that included hepatopathy, bifid uvula, malignant hyperthermia, hypogonadotropic hypogonadism, growth retardation, hypoglycemia, myopathy, dilated cardiomyopathy, and cardiac arrest.
Methods: Homozygosity mapping followed by whole-exome sequencing was used to identify a mutation in the gene for phosphoglucomutase 1 (PGM1) in two siblings. Sequencing identified additional mutations in 15 other families. Phosphoglucomutase 1 enzyme activity was assayed on cell extracts. Analyses of glycosylation efficiency and quantitative studies of sugar metabolites were performed. Galactose supplementation in fibroblast cultures and dietary supplementation in the patients were studied to determine the effect on glycosylation.
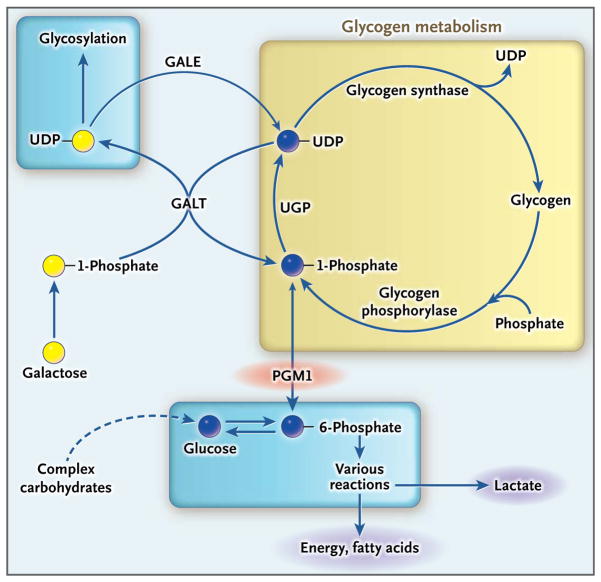
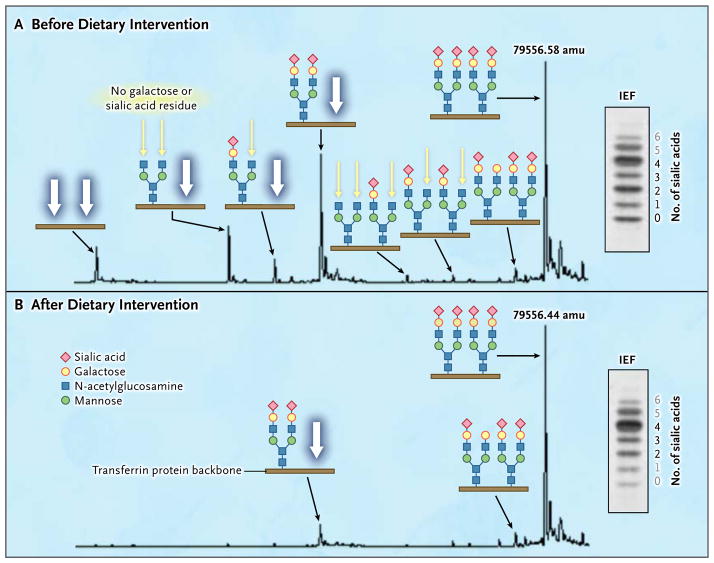
Results: Phosphoglucomutase 1 enzyme activity was markedly diminished in all patients. Mass spectrometry of transferrin showed a loss of complete N-glycans and the presence of truncated glycans lacking galactose. Fibroblasts supplemented with galactose showed restoration of protein glycosylation and no evidence of glycogen accumulation. Dietary supplementation with galactose in six patients resulted in changes suggestive of clinical improvement. A new screening test showed good discrimination between patients and controls.
Conclusions: Phosphoglucomutase 1 deficiency, previously identified as a glycogenosis, is also a congenital disorder of glycosylation. Supplementation with galactose leads to biochemical improvement in indexes of glycosylation in cells and patients, and supplementation with complex carbohydrates stabilizes blood glucose. A new screening test has been developed but has not yet been validated. (Funded by the Netherlands Organization for Scientific Research and others.).
Figures
Comment in
-
Multiple phenotypes in phosphoglucomutase 1 deficiency.N Engl J Med. 2014 May 22;370(21):2051-2. doi: 10.1056/NEJMc1403446. N Engl J Med. 2014. PMID: 24849093 No abstract available.
-
Multiple phenotypes in phosphoglucomutase 1 deficiency.N Engl J Med. 2014 May 22;370(21):2051. doi: 10.1056/NEJMc1403446. N Engl J Med. 2014. PMID: 24849094 No abstract available.
Similar articles
-
Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG.Mol Genet Metab. 2014 Aug;112(4):275-9. doi: 10.1016/j.ymgme.2014.06.002. Epub 2014 Jun 21. Mol Genet Metab. 2014. PMID: 24997537 Free PMC article. Review.
-
Phosphoglucomutase-1 deficiency: Early presentation, metabolic management and detection in neonatal blood spots.Mol Genet Metab. 2020 Sep-Oct;131(1-2):135-146. doi: 10.1016/j.ymgme.2020.08.003. Epub 2020 Sep 17. Mol Genet Metab. 2020. PMID: 33342467
-
Intact transferrin and total plasma glycoprofiling for diagnosis and therapy monitoring in phosphoglucomutase-I deficiency.Transl Res. 2018 Sep;199:62-76. doi: 10.1016/j.trsl.2018.04.008. Epub 2018 May 10. Transl Res. 2018. PMID: 30048639 Free PMC article.
-
Phosphoglucomutase-1 deficiency: Intrafamilial clinical variability and common secondary adrenal insufficiency.Am J Med Genet A. 2015 Dec;167A(12):3139-43. doi: 10.1002/ajmg.a.37294. Epub 2015 Aug 19. Am J Med Genet A. 2015. PMID: 26768186
-
Mutations in hereditary phosphoglucomutase 1 deficiency map to key regions of enzyme structure and function.J Inherit Metab Dis. 2015 Mar;38(2):243-56. doi: 10.1007/s10545-014-9757-9. Epub 2014 Aug 29. J Inherit Metab Dis. 2015. PMID: 25168163 Review.
Cited by
-
Skeletal muscle disorders of glycogenolysis and glycolysis.Nat Rev Neurol. 2016 Jul;12(7):393-402. doi: 10.1038/nrneurol.2016.75. Epub 2016 May 27. Nat Rev Neurol. 2016. PMID: 27231184 Review.
-
Liver glucose metabolism in humans.Biosci Rep. 2016 Nov 29;36(6):e00416. doi: 10.1042/BSR20160385. Print 2016 Dec. Biosci Rep. 2016. PMID: 27707936 Free PMC article. Review.
-
Identification and characterization of differentially expressed exosomal microRNAs in bovine milk infected with Staphylococcus aureus.BMC Genomics. 2019 Dec 5;20(1):934. doi: 10.1186/s12864-019-6338-1. BMC Genomics. 2019. PMID: 31805863 Free PMC article.
-
A Double-Blind, Placebo-Controlled, Crossover Study of Magnesium Supplementation in Patients with XMEN Disease.J Clin Immunol. 2022 Jan;42(1):108-118. doi: 10.1007/s10875-021-01137-w. Epub 2021 Oct 16. J Clin Immunol. 2022. PMID: 34655400 Free PMC article. Clinical Trial.
-
Genetic characteristics of patients with congenital hyperinsulinism.Curr Opin Pediatr. 2018 Aug;30(4):568-575. doi: 10.1097/MOP.0000000000000645. Curr Opin Pediatr. 2018. PMID: 29750770 Free PMC article. Review.
References
-
- Quick CB, Fisher RA, Harris H. A kinetic study of the isozymes determined by the three human phosphoglucomutase loci PGM1, PGM2, and PGM3. Eur J Biochem. 1974;42:511–7. - PubMed
-
- Stojkovic T, Vissing J, Petit F, et al. Muscle glycogenosis due to phosphoglucomutase 1 deficiency. N Engl J Med. 2009;361:425–7. - PubMed
-
- de Jong G, van Noort WL, van Eijk HG. Optimized separation and quantitation of serum and cerebrospinal fluid transferrin subfractions defined by differences in iron saturation or glycan composition. Adv Exp Med Biol. 1994;356:51–9. - PubMed
-
- Mandato C, Brive L, Miura Y, et al. Cryptogenic liver disease in four children: a novel congenital disorder of glycosylation. Pediatr Res. 2006;59:293–8. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous