

Branching enzyme deficiency: expanding the clinical spectrum
- PMID: 24248152
- PMCID: PMC6148323
- DOI: 10.1001/jamaneurol.2013.4888
Branching enzyme deficiency: expanding the clinical spectrum
Abstract
Importance: The neuromuscular presentation of glycogen branching enzyme deficiency includes a severe infantile form and a late-onset variant known as adult polyglucosan body disease. Herein, we describe 2 patients with adult acute onset of fluctuating neurological signs and brain magnetic resonance imaging lesions simulating multiple sclerosis. A better definition of this new clinical entity is needed to facilitate diagnosis.
Objectives: To describe the clinical presentation and progression of a new intermediate variant of glycogen branching enzyme deficiency and to discuss genotype-phenotype correlations.
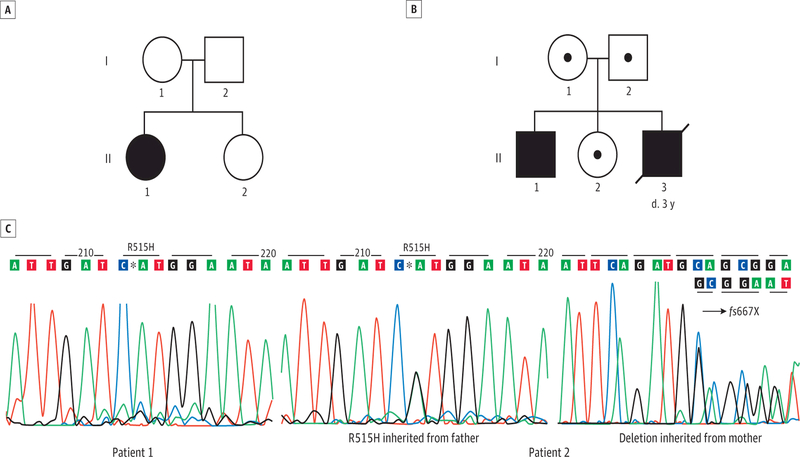
Design, setting, and participants: Clinical, biochemical, morphological, and molecular study of 2 patients followed up for 6 years and 8 years at academic medical centers. The participants were 2 patients of non-Ashkenazi descent with adult acute onset of neurological signs initially diagnosed as multiple sclerosis.
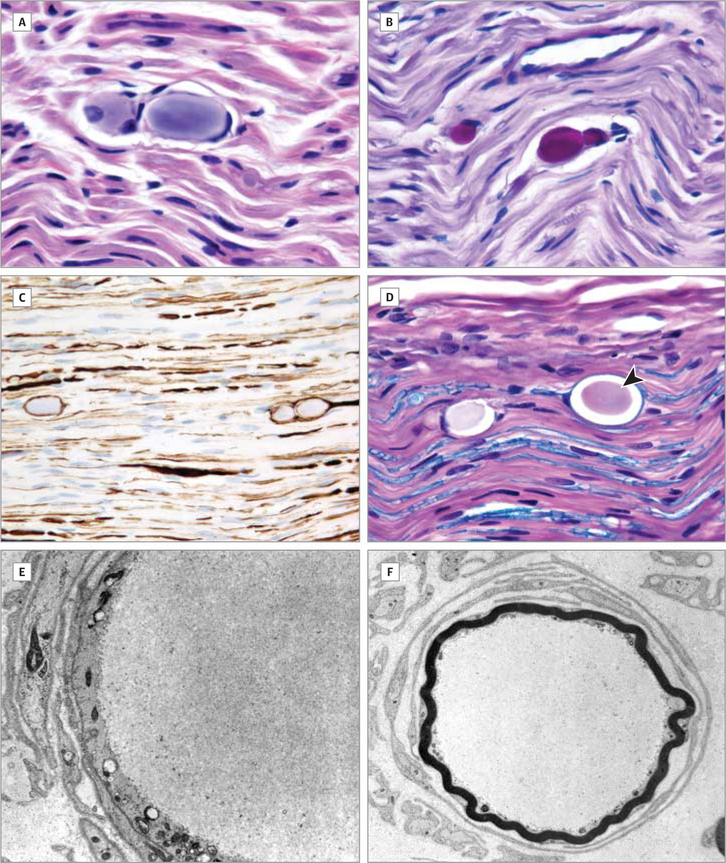
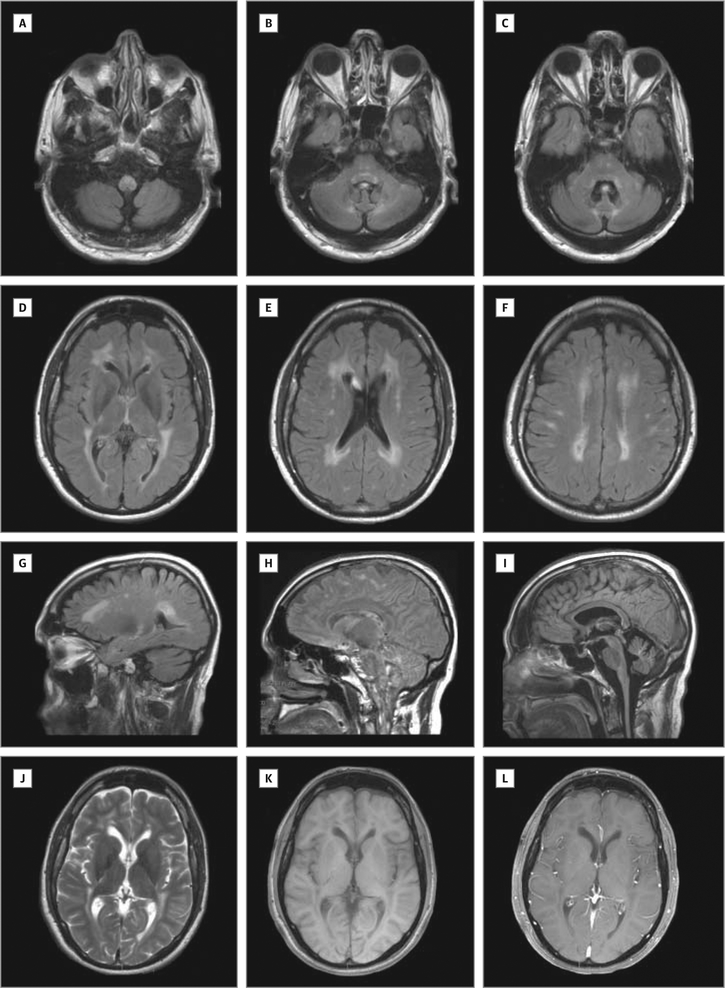
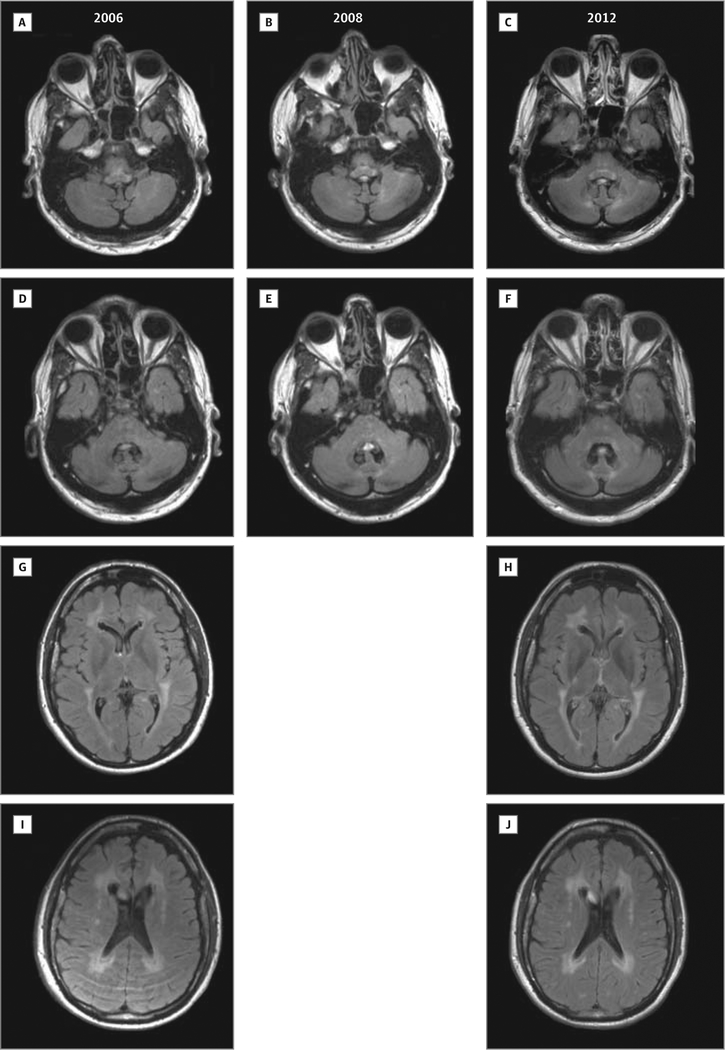
Main outcomes and measures: Clinical course, muscle and nerve morphology, longitudinal study of brain magnetic resonance imaging, and glycogen branching enzyme activity and GBE1 molecular analysis.
Results: Molecular analysis showed that one patient was homozygous (c.1544G>A) and the other patient was compound heterozygous (c.1544G>A and c.1961-1962delCA) for GBE1 mutations. Residual glycogen branching enzyme activity was 16% and 30% of normal in leukocytes. Both patients manifested acute episodes of transient neurological symptoms, and neurological impairment was mild at age 45 years and 53 years. Brain magnetic resonance imaging revealed nonprogressive white matter lesions and spinocerebellar atrophy similar to typical adult polyglucosan body disease.
Conclusions and relevance: GBE1 mutations can cause an early adult-onset relapsing-remitting form of polyglucosan body disease distinct from adult polyglucosan body disease in several ways, including younger age at onset, history of infantile liver involvement, and subacute and remitting course simulating multiple sclerosis. This should orient neurologists toward the correct diagnosis.
Conflict of interest statement
Figures
Similar articles
-
Neuropathological study of skeletal muscle, heart, liver, and brain in a neonatal form of glycogen storage disease type IV associated with a new mutation in GBE1 gene.J Inherit Metab Dis. 2009 Dec;32 Suppl 1:S161-8. doi: 10.1007/s10545-009-1134-8. Epub 2009 Apr 8. J Inherit Metab Dis. 2009. PMID: 19357989
-
Structural basis of glycogen branching enzyme deficiency and pharmacologic rescue by rational peptide design.Hum Mol Genet. 2015 Oct 15;24(20):5667-76. doi: 10.1093/hmg/ddv280. Epub 2015 Jul 21. Hum Mol Genet. 2015. PMID: 26199317 Free PMC article.
-
A novel mouse model that recapitulates adult-onset glycogenosis type 4.Hum Mol Genet. 2015 Dec 1;24(23):6801-10. doi: 10.1093/hmg/ddv385. Epub 2015 Sep 18. Hum Mol Genet. 2015. PMID: 26385640 Free PMC article.
-
Diffuse reticuloendothelial system involvement in type IV glycogen storage disease with a novel GBE1 mutation: a case report and review.Hum Pathol. 2012 Jun;43(6):943-51. doi: 10.1016/j.humpath.2011.10.001. Epub 2012 Feb 2. Hum Pathol. 2012. PMID: 22305237 Review.
-
GBE1-related disorders: Adult polyglucosan body disease and its neuromuscular phenotypes.J Inherit Metab Dis. 2021 May;44(3):534-543. doi: 10.1002/jimd.12325. Epub 2020 Nov 13. J Inherit Metab Dis. 2021. PMID: 33141444 Review.
Cited by
-
Frequent misdiagnosis of adult polyglucosan body disease.J Neurol. 2015 Oct;262(10):2346-51. doi: 10.1007/s00415-015-7859-4. Epub 2015 Jul 21. J Neurol. 2015. PMID: 26194201
-
Preclinical Development of New Therapy for Glycogen Storage Diseases.Curr Gene Ther. 2015;15(4):338-47. doi: 10.2174/1566523215666150630132253. Curr Gene Ther. 2015. PMID: 26122079 Free PMC article. Review.
-
A Broad Characterization of Glycogen Storage Disease IV Patients: A Clinical, Genetic, and Histopathological Study.Biomedicines. 2023 Jan 26;11(2):363. doi: 10.3390/biomedicines11020363. Biomedicines. 2023. PMID: 36830903 Free PMC article.
-
Neurological glycogen storage diseases and emerging therapeutics.Neurotherapeutics. 2024 Sep;21(5):e00446. doi: 10.1016/j.neurot.2024.e00446. Epub 2024 Sep 14. Neurotherapeutics. 2024. PMID: 39277505 Free PMC article. Review.
-
Neural correlates of adaptive working memory training in a glycogen storage disease type-IV patient.Ann Clin Transl Neurol. 2017 Feb 23;4(3):217-222. doi: 10.1002/acn3.394. eCollection 2017 Mar. Ann Clin Transl Neurol. 2017. PMID: 28275655 Free PMC article.
References
-
- Magoulas PL, El-Hattab AW. Glycogen storage disease type IV [Internet] In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews. Seattle: University of Washington; 1993–2013. - PubMed
-
- Schröder JM, May R, Shin YS, Sigmund M, Nase-Hüppmeier S. Juvenile hereditary polyglucosan body disease with complete branching enzyme deficiency (type IV glycogenosis). Acta Neuropathol. 1993;85(4):419–430. - PubMed
-
- Bruno C, Servidei S, Shanske S, et al. Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann Neurol. 1993;33(1):88–93. - PubMed
-
- Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV. Curr Mol Med. 2002;2(2):177–188. - PubMed
-
- Bruno C, van Diggelen OP, Cassandrini D, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. 2004;63(6):1053–1058. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
