

A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations
- PMID: 23758768
- PMCID: PMC3718741
- DOI: 10.1186/1750-1172-8-84
A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations
Abstract
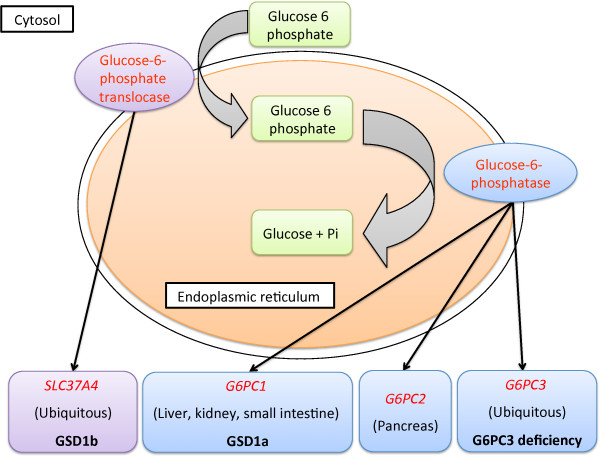
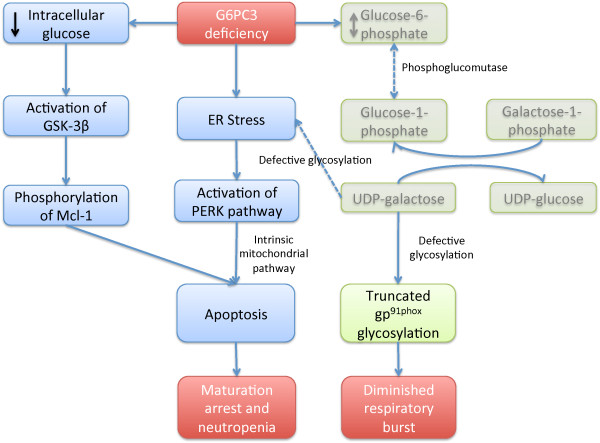
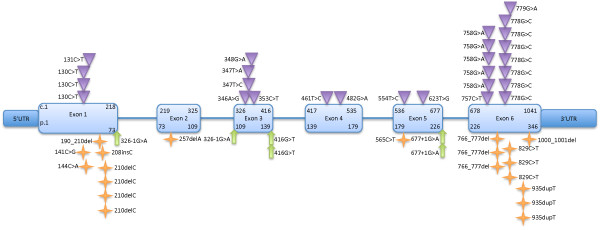
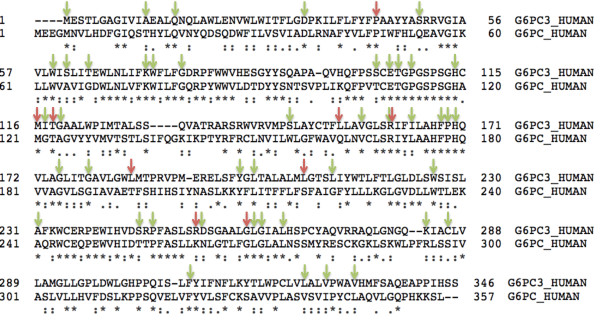
The G6PC3 gene encodes the ubiquitously expressed glucose-6-phosphatase enzyme (G-6-Pase β or G-6-Pase 3 or G6PC3). Bi-allelic G6PC3 mutations cause a multi-system autosomal recessive disorder of G6PC3 deficiency (also called severe congenital neutropenia type 4, MIM 612541). To date, at least 57 patients with G6PC3 deficiency have been described in the literature.G6PC3 deficiency is characterized by severe congenital neutropenia, recurrent bacterial infections, intermittent thrombocytopenia in many patients, a prominent superficial venous pattern and a high incidence of congenital cardiac defects and uro-genital anomalies. The phenotypic spectrum of the condition is wide and includes rare manifestations such as maturation arrest of the myeloid lineage, a normocellular bone marrow, myelokathexis, lymphopaenia, thymic hypoplasia, inflammatory bowel disease, primary pulmonary hypertension, endocrine abnormalities, growth retardation, minor facial dysmorphism, skeletal and integument anomalies amongst others. Dursun syndrome is part of this extended spectrum. G6PC3 deficiency can also result in isolated non-syndromic severe neutropenia. G6PC3 mutations in result in reduced enzyme activity, endoplasmic reticulum stress response, increased rates of apoptosis of affected cells and dysfunction of neutrophil activity.In this review we demonstrate that loss of function in missense G6PC3 mutations likely results from decreased enzyme stability. The condition can be diagnosed by sequencing the G6PC3 gene. A number of G6PC3 founder mutations are known in various populations and a possible genotype-phenotype relationship also exists. G6PC3 deficiency should be considered as part of the differential diagnoses in any patient with unexplained congenital neutropenia.Treatment with G-CSF leads to improvement in neutrophil numbers, prevents infections and improves quality of life. Mildly affected patients can be managed with prophylactic antibiotics. Untreated G6PC3 deficiency can be fatal. Echocardiogram, renal and pelvic ultrasound scans should be performed in all cases of suspected or confirmed G6PC3 deficiency. Routine assessment should include biochemical profile, growth profile and monitoring for development of varicose veins or venous ulcers.
Figures
Similar articles
-
Three patients with glucose-6 phosphatase catalytic subunit 3 deficiency.J Pediatr Endocrinol Metab. 2020 Jul 28;33(7):957-961. doi: 10.1515/jpem-2019-0541. J Pediatr Endocrinol Metab. 2020. PMID: 32623377
-
Mutations in the G6PC3 gene cause Dursun syndrome.Am J Med Genet A. 2010 Oct;152A(10):2609-11. doi: 10.1002/ajmg.a.33615. Am J Med Genet A. 2010. PMID: 20799326
-
G6PC3 mutations cause non-syndromic severe congenital neutropenia.Mol Genet Metab. 2013 Feb;108(2):138-41. doi: 10.1016/j.ymgme.2012.12.001. Epub 2012 Dec 21. Mol Genet Metab. 2013. PMID: 23298686
-
Novel G6PC3 Mutations in Patients with Congenital Neutropenia: Case Reports and Review of the Literature.Endocr Metab Immune Disord Drug Targets. 2021;21(9):1660-1668. doi: 10.2174/1871530321666210616110631. Endocr Metab Immune Disord Drug Targets. 2021. PMID: 34137364 Review.
-
The molecular basis of type 1 glycogen storage diseases.Curr Mol Med. 2001 Mar;1(1):25-44. doi: 10.2174/1566524013364112. Curr Mol Med. 2001. PMID: 11899241 Review.
Cited by
-
WHIM Syndrome: from Pathogenesis Towards Personalized Medicine and Cure.J Clin Immunol. 2019 Aug;39(6):532-556. doi: 10.1007/s10875-019-00665-w. Epub 2019 Jul 16. J Clin Immunol. 2019. PMID: 31313072 Free PMC article. Review.
-
Molecular and clinical characterization of a founder mutation causing G6PC3 deficiency.medRxiv [Preprint]. 2024 May 14:2024.05.13.24307299. doi: 10.1101/2024.05.13.24307299. medRxiv. 2024. Update in: J Clin Immunol. 2024 Dec 4;45(1):53. doi: 10.1007/s10875-024-01836-0. PMID: 38798393 Free PMC article. Updated. Preprint.
-
Esophageal Stricture Secondary to Candidiasis in a Child with Glycogen Storage Disease 1b.Pediatr Gastroenterol Hepatol Nutr. 2016 Mar;19(1):71-5. doi: 10.5223/pghn.2016.19.1.71. Epub 2016 Mar 22. Pediatr Gastroenterol Hepatol Nutr. 2016. PMID: 27066451 Free PMC article.
-
Functional characteristics of circulating granulocytes in severe congenital neutropenia caused by ELANE mutations.BMC Pediatr. 2019 Jun 8;19(1):189. doi: 10.1186/s12887-019-1556-x. BMC Pediatr. 2019. PMID: 31176364 Free PMC article.
-
SLGT2 Inhibitor Rescues Myelopoiesis in G6PC3 Deficiency.J Clin Immunol. 2022 Nov;42(8):1653-1659. doi: 10.1007/s10875-022-01323-4. Epub 2022 Jul 15. J Clin Immunol. 2022. PMID: 35838821
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
