

Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome
- PMID: 23687348
- PMCID: PMC3717725
- DOI: 10.1136/jmedgenet-2012-101405
Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome
Abstract
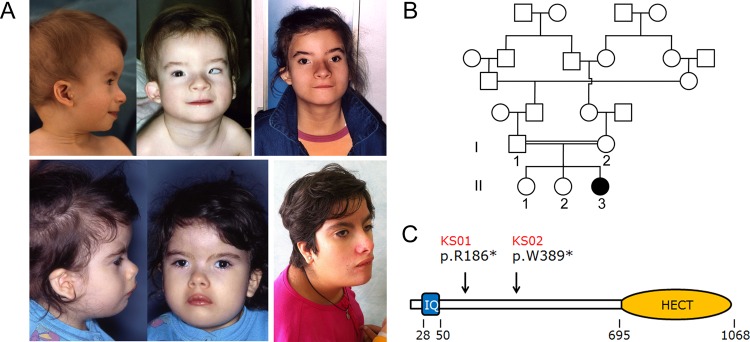
Background: Kaufman oculocerebrofacial syndrome (KOS) is a developmental disorder characterised by reduced growth, microcephaly, ocular anomalies (microcornea, strabismus, myopia, and pale optic disk), distinctive facial features (narrow palpebral fissures, telecanthus, sparse and laterally broad eyebrows, preauricular tags, and micrognathia), mental retardation, and generalised hypotonia. KOS is a rare, possibly underestimated condition, with fewer than 10 cases reported to date. Here we investigate the molecular cause underlying KOS.
Methods: An exome sequencing approach was used on a single affected individual of an Italian consanguineous family coupled with mutation scanning using Sanger sequencing on a second unrelated subject with clinical features fitting the disorder.
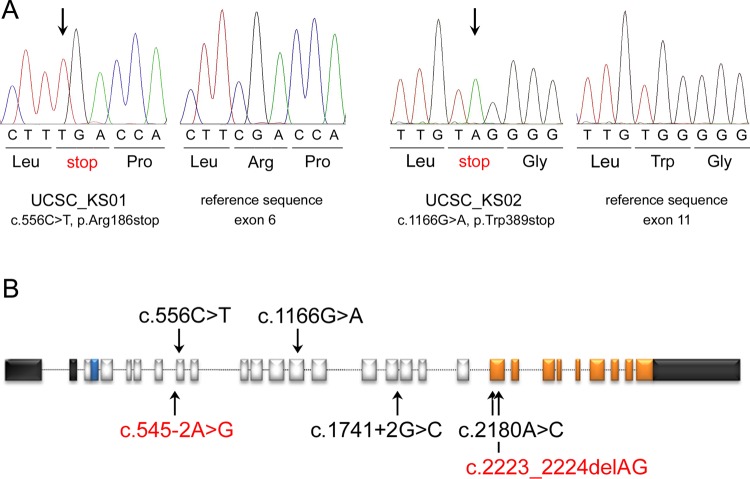
Results: Exome sequencing was able to identify homozygosity for a novel truncating mutation (c.556C>T, p.Arg186stop) in UBE3B, which encodes a widely expressed HECT (homologous to the E6-AP carboxyl terminus) domain E3 ubiquitin-protein ligase. Homozygosity for a different nonsense lesion affecting the gene (c.1166G>A, p.Trp389stop) was documented in the second affected subject, supporting the recessive mode of inheritance of the disorder. Mutation scanning of the entire UBE3B coding sequence on a selected cohort of subjects with features overlapping, in part, those recurring in KOS did not reveal disease-causing mutations, suggesting phenotypic homogeneity of UBE3B lesions.
Discussion: Our data provide evidence that KOS is caused by UBE3B loss of function, and further demonstrate the impact of misregulation of protein ubiquitination on development and growth. The available clinical records, including those referring to four UBE3B mutation-positive subjects recently described as belonging to a previously unreported entity, which fits KOS, document the clinical homogeneity of this disorder.
Keywords: Clinical genetics; Developmental; Diagnosis; Genome-wide; Molecular genetics.
Figures
Similar articles
-
Expanding the clinical and mutational spectrum of Kaufman oculocerebrofacial syndrome with biallelic UBE3B mutations.Hum Genet. 2014 Jul;133(7):939-49. doi: 10.1007/s00439-014-1436-2. Epub 2014 Mar 11. Hum Genet. 2014. PMID: 24615390
-
Novel UBE3B mutations: report of eight patients with Kaufman oculocerebrofacial syndrome with additional clinical findings from a highly consanguineous population.Clin Dysmorphol. 2024 Apr 1;33(2):55-62. doi: 10.1097/MCD.0000000000000486. Epub 2024 Feb 15. Clin Dysmorphol. 2024. PMID: 38410982
-
Kaufman oculocerebrofacial syndrome in sisters with novel compound heterozygous mutation in UBE3B.Am J Med Genet A. 2015 Mar;167A(3):657-63. doi: 10.1002/ajmg.a.36944. Am J Med Genet A. 2015. PMID: 25691420
-
Further phenotypic characterization of Kaufman oculocerebrofacial syndrome: report of five new cases and literature review.Clin Dysmorphol. 2019 Oct;28(4):175-183. doi: 10.1097/MCD.0000000000000282. Clin Dysmorphol. 2019. PMID: 31162149 Review.
-
A new case of Kaufman Oculocerebrofacial syndrome caused by two splicing variants in UBE3B and review of the literature.Clin Genet. 2023 Mar;103(3):377-379. doi: 10.1111/cge.14270. Epub 2022 Dec 1. Clin Genet. 2023. PMID: 36444497 Review. No abstract available.
Cited by
-
UBE3B promotes breast cancer progression by antagonizing HIF-2α degradation.Oncogene. 2023 Nov;42(46):3394-3406. doi: 10.1038/s41388-023-02842-z. Epub 2023 Oct 2. Oncogene. 2023. PMID: 37783786
-
E3 Ubiquitin Ligases in Neurological Diseases: Focus on Gigaxonin and Autophagy.Front Physiol. 2020 Oct 22;11:1022. doi: 10.3389/fphys.2020.01022. eCollection 2020. Front Physiol. 2020. PMID: 33192535 Free PMC article. Review.
-
Rare copy number variations containing genes involved in RASopathies: deletion of SHOC2 and duplication of PTPN11.Mol Cytogenet. 2014 Apr 16;7:28. doi: 10.1186/1755-8166-7-28. eCollection 2014. Mol Cytogenet. 2014. PMID: 24739123 Free PMC article.
-
A combined genome-wide association and molecular study of age-related hearing loss in H. sapiens.BMC Med. 2021 Dec 1;19(1):302. doi: 10.1186/s12916-021-02169-0. BMC Med. 2021. PMID: 34847940 Free PMC article.
-
Expanding the clinical and mutational spectrum of Kaufman oculocerebrofacial syndrome with biallelic UBE3B mutations.Hum Genet. 2014 Jul;133(7):939-49. doi: 10.1007/s00439-014-1436-2. Epub 2014 Mar 11. Hum Genet. 2014. PMID: 24615390
References
-
- Kaufman R, Rimoin DL, Prensky AL, Sly WS. An oculocerebrofacial syndrome. Birth Defects Orig Art Ser 1971;7:135–8 - PubMed
-
- Jurenka SB, Evans J. Kaufman oculocerebrofacial syndrome: case report. Am J Med Genet 1979;3:15–19 - PubMed
-
- Garcia-Cruz D, Arreola R, Sanchez-Corona J, Garcia-Cruz O, Renteria R, Villar V, Gonzalez ME, Vargas-Moyeda E, Cantu JM. Kaufman oculocerebrofacial syndrome: a corroborative report. Dysmorph Clin Genet 1988;1:152–4
-
- Figuera LE, Garcia-Cruz D, Ramirez-Duenas ML, Rivera-Robles V, Cantu JM. Kaufman oculocerebrofacial syndrome: report of two new cases and further delineation. Clin Genet 1993;44:98–101 - PubMed
-
- Verloes A, Bremond-Grignac D, Isidor B, David A, Baumann C, Leroy MA, Stevens R, Gillerot Y, Heron D, Heron B, Benzacken B, Lacombe D, Brunner H, Bitoun P. Blepharophimosis-mental retardation (BMR) syndromes: a proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A 2006;140:1285–96 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical